
Abstract
Tea plants are subjected to multiple stresses during growth, development, and postharvest processing, which affects levels of secondary metabolites in leaves and influences tea functional properties and quality. Most studies on secondary metabolism in tea have focused on gene, protein, and metabolite levels, whereas upstream regulatory mechanisms remain unclear. In this review, we exemplify DNA methylation and histone acetylation, summarize the important regulatory effects that epigenetic modifications have on plant secondary metabolism, and discuss feasible research strategies to elucidate the underlying specific epigenetic mechanisms of secondary metabolism regulation in tea. This information will help researchers investigate the epigenetic regulation of secondary metabolism in tea, providing key epigenetic data that can be used for future tea genetic breeding.
Similar content being viewed by others


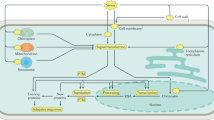
Introduction
Tea (Camellia sinensis) is an important economic crop in China. Indeed, tea plants have been cultivated and used in China for thousands of years. Tea is currently second only to water as the most important beverage worldwide1. Compared with other plants, tea contains unique secondary metabolites, including catechins, amino acids (mainly L-theanine), caffeine, and aroma compounds, giving it unique color, aroma, and flavor qualities while also influencing human health2,3. Hence, there has been increasing interest in research on the formation of compounds related to tea function and quality. These compounds are mainly formed during growth (i.e., the preharvest stage) and the processing (i.e., the postharvest stage)4,5,6. Previous studies have explored the formation of function- or quality-related metabolites in enzymatic and nonenzymatic (i.e., chemical) reactions1,4, and the formation of tea metabolites from a plant biology perspective has also been investigated. Thus, secondary metabolism and associated regulatory processes in tea plants are important research topics.
Environmental stresses affect the formation of secondary metabolites in plants via physiological and biochemical mechanisms in adaptation to environmental conditions and resistance to adverse effects caused by external stresses7. Most of the traditional theories regarding plant metabolic responses to stress are based on analyses of plant stress tolerance. In general, tea is exposed to multiple stresses during the preharvest and postharvest stages of production, which can induce enrichment of secondary metabolites closely related to quality or function3,4. For example, shading is commonly used in the preharvest stage to change the light conditions to increase the contents of flavor-related compounds, including amino acids and aromatics, while decreasing the abundance of polyphenols such as catechins (reducing bitterness)5. Therefore, a thorough characterization of the mechanism by which these stressors induce the formation of secondary metabolites in tea and the application of the mechanism to improve the quality of tea components may lead to the development of new methods for safely enhancing tea quality.
To date, studies on the mechanism underlying stress-induced secondary metabolites in tea have mainly focused on associated genes, proteins, and metabolites, especially the stress-induced increase in target metabolite contents, which is mainly due to upregulated expression of key synthesis-related genes. For example, exposure to low temperatures and continuous wounding synergistically increase indole, jasmine lactone, and (E)-nerolidol contents in oolong tea mainly because of stress-induced expression of CsTSB2, CsLOX1, and CsNES, which are key genes mediating the synthesis of these aroma compounds8,9,10,11,12. However, regarding the upstream regulatory mechanisms, there are only a few reports describing the involvement of some plant hormones and transcription factors. Specifically, upstream regulation of the formation of tea aroma compounds synergistically induced by low temperatures and continuous wounding is mainly related to jasmonic acid and the transcription factor CsMYC211. Other factors involved in upstream regulatory activities, such as epigenetic regulators, remain poorly understood. Epigenetic regulation in various plants is closely related to stress and secondary metabolite production13,14,15,16,17,18, but there is relatively little research with regard to tea19,20. Accordingly, there is an urgent need for studies on the epigenetic regulation of tea quality (Fig. 1), as the paucity of research on the upstream signaling mechanisms of secondary metabolism has not only limited molecular analyses of tea but has also restricted the development of novel methods for improving its quality.

As tea is a nonmodel plant species that lacks a well-established genetic transformation system, there has been relatively little epigenetic-related research on this plant. Thus, researchers are faced with many challenges when conducting epigenetic studies on the regulation of secondary metabolism in tea. For example, it is unclear how to verify whether epigenetics is involved in the regulation of secondary metabolism in tea and what the main epigenetic factors involved in the regulation are. In this review, we explore research strategies for investigating the epigenetic control of secondary metabolism in tea on the basis of the genetic characteristics of these plants, focusing on the following two major epigenetic modifications as examples: DNA methylation and histone deacetylation. First, we review epigenetic regulation of plant secondary metabolism and explore the possibility that DNA methylation and histone acetylation are involved in the upstream regulation of key genes of tea quality-related metabolites in tea induced by stress. Second, possible regulatory patterns of DNA methylation and histone acetylation are summarized. Third, a feasible strategy for studying epigenetic regulation of plant secondary metabolism in tea is proposed. The main objective of this review is to summarize research strategies for studying the epigenetic regulation of tea secondary metabolism and providing key epigenetic factors that can be used for future tea genetic breeding.
DNA methylation and histone acetylation may be the main upstream regulators of the expression of key genes influencing stress-induced tea quality-related metabolism
In plants, DNA methylation is primarily responsible for regulating physiological and biochemical processes such as gene expression, cell differentiation, metabolism, and stress responses21. For example, when plants are stimulated by external stresses, the genomic DNA methylation status changes (i.e., increases or decreases) to modulate chromatin structure and expression of genes related to responses to environmental stimuli22. Histone modifications, including acetylation, methylation, phosphorylation, and ubiquitination, involve covalent changes to histone amino acids. Histone acetylation relaxes nucleosome structures, making DNA more receptive to transcription factors that activate specific genes; in contrast, histone deacetylation and partially methylated histone sites (e.g., K9 and K27) tighten chromatin and inhibit transcription23,24. Histone acetylation is closely related to gene silencing, seed germination, morphogenesis, and stress responses25, and studies have confirmed that many types of epigenetic changes regulate various plant growth and developmental processes, including flowering, root/stem cell maintenance, hypocotyl elongation, embryogenesis, seed germination, and responses to biotic and abiotic stresses26,27. However, in tea plants, epigenetic regulation of plant secondary metabolite formation has not been thoroughly studied. Here, we focus on the role of DNA methylation and histone acetylation modifications on the regulation of secondary metabolites in tea under stress conditions and propose a feasible research strategy.
Compared with epigenetic studies of model plants, progress in nonmodel plants is relatively slow, especially because most nonmodel plants exhibit characteristics of a long growth cycle and abundant population, which determine the uniqueness and innovation of their epigenetic research. With the development of epigenetic research methods over the past two decades, significant progress has been made in research on epigenetic regulation of secondary metabolism, which has improved our understanding of epigenetic regulation of secondary metabolism in nonmodel plants. In recent years, epigenetic modifications have been found to be involved in secondary metabolism regulation in many plants (Table 1). Epigenetic involvement is determined by detecting metabolite contents and gene expression changes after inhibitor treatment28,29,30. Restriction enzyme digestion and bisulfite sequencing are also used to analyze methylation level changes of target genes and the specific mechanism of epigenetic modification regulating secondary metabolism13,14,16,17,18,31,32. Among these studies, DNA methylation and histone acetylation modification are particularly notable.
As a typical nonmodel plant, tea plants are rich in secondary metabolites. Therefore, it is important to investigate the regulatory role of epigenetic modification in secondary metabolite formation in tea plants. Despite relatively recent molecular investigations in tea, researchers have clarified the effects of epigenetic modifications on specific aspects (Table 2). To elucidate the regulatory role of epigenetics in different aspects of tea plants, models have been selected according to research objectives. For example, from the perspective of genome-wide and overall methylation levels, it was found that DNA methylation and histone acetylation are involved in the evolution and stress response of tea plants, revealing the genetic basis and providing new insight into the mechanism of flavor substance formation and quality regulation of tea33,34,35,36,37. From the perspective of transcriptomics, results of analysis of the biological information of epigenetic modification and epigenetic regulatory factors in tea plants have suggested that DNA methylation plays an important role in the regulation of stress response and growth and development38,39. From the perspective of proteomics, elucidating the regulation of specific modification sites with regard to the formation of secondary metabolites has provided important insight into the regulatory role of lysine acetylation in secondary metabolism in tea plants40,41. Based on changes in the DNA methylation level and histone modification of the target genes, the regulatory factors involved in the regulation of tea processing, low temperature and long illumination have been identified, clarifying the specific regulatory mechanism of the formation of secondary metabolites19,20,42,43,44. These results suggest that epigenetic modifications help to regulate stress responses and secondary metabolite biosynthesis in tea. Accordingly, the stress-induced formation of secondary metabolites in tea may be related to the regulation of DNA methylation and histone acetylation.
Studies on the regulation of DNA methylation and histone acetylation of key genes mediating stress-induced tea quality-related metabolism
Many studies have demonstrated that epigenetic modifications influence plant stress responses, providing new data and directions for epigenetic research45,46. Subsequent studies have found a correlation between epigenetic modifications and secondary metabolite contents and quality47. In tea, some secondary metabolites are formed in the postharvest stage; this is in contrast with the preharvest stage, during which plants are damaged by various stresses1. Secondary metabolites are closely associated with tea quality. Hence, studying the epigenetic status of tea plants under different stress conditions, resolving the epigenetic modifications induced by stresses, and revealing key factors influencing tea quality will provide the basis for future investigations of the molecular mechanisms underlying processes affecting tea quality.
Nevertheless, epigenetic-based research on tea lags behind that of model plants because of its long growth cycle, large genomes, and varietal diversity. For example, unlike Arabidopsis, it is difficult to obtain tea mutants and a stable genetic transformation system. Accordingly, generating accurate and valid in vivo experimental data when functionally characterizing target genes is challenging, which hinders exploration of the consequences of epigenetic modifications on secondary metabolites in tea. Furthermore, epigenetic regulation in tea exposed to abiotic stress is a dynamic process that requires sophisticated experimental techniques for analysis [e.g., polyphenols may affect reverse crosslinking during chromatin immunoprecipitation (ChIP) experiments or cause DNA damage48]. Thus, analyzing the epigenetic mechanism in tea under stress conditions is very difficult. In general, determining whether DNA methylation and histone deacetylation are involved in the regulation of secondary metabolism under these conditions is critical and should be the first step in the study of stress-induced epigenetic modifications in tea. This can be achieved in the following three ways (Fig. 2). (1) Samples are treated with DNA methylation and histone acetylation inhibitors, after which the secondary metabolite contents in control and treatment groups are analyzed to obtain direct evidence of a correlation in vivo. If there is no difference in secondary metabolite contents between the groups, DNA methylation and histone acetylation are not involved in regulating the secondary metabolism related to this process. In contrast, significant differences in secondary metabolite contents between the groups suggest that DNA methylation and histone acetylation help to regulate the secondary metabolism related to this process. (2) Secondary metabolite contents in tea are determined under diverse stress conditions49, and levels of DNA methylation and histone acetylation of the synthase genes involved in the regulation of secondary metabolites are analyzed. (3) Transcription of DNA methyltransferase, DNA demethylase, histone acetyltransferase (HAT), and histone deacetylase (HDAC) genes in tea are quantified under various stress conditions, and the data are analyzed via nontargeted correlation analyses. Significant differences in the expression of genes related to DNA methylation and histone acetylation under control and stress conditions suggest that both are involved in the secondary metabolism that regulates this process. If there is no significant difference between sample groups, differences in the abundance of the encoded DNA methylation-related and histone acetylation-related enzymes are determined, and significant differences in enzyme levels between control and stress conditions indicate that DNA methylation and histone acetylation are involved in the secondary metabolism that regulates this process. If there is no significant difference, the binding of HDACs to target genes is evaluated. If the ability of HDACs to bind to the target genes varies between control and stress conditions, histone acetylation is involved in regulating the secondary metabolism of this process, whereas a lack of a significant difference indicates a lack of involvement.

a In vivo evidence of inhibitor treatment. b Analysis of target metabolites under stress. c Analysis of transcription level of epigenetic factors under stress. YES or NO denotes judging whether DNA methylation and histone acetylation are involved in the regulation of secondary metabolism in tea plants. HOW represents the mechanism of DNA methylation and histone acetylation involvement in the regulation of secondary metabolism in tea plants. TFs transcription factors, HDACs histone deacetylases, MBDs methyl-binding domain protein, ChIP chromatin immunoprecipitation, EMSA electrophoretic mobility shift assay, Y2H yeast two-hybrid, BiFC bimolecular fluorescence complementation, Co-IP coimmunoprecipitation.
In the first determination method (Fig. 2, a route), diverse DNA methylation and histone acetylation inhibitors are available for analyses (Table 3). Regarding DNA methylation, tea leaves can be treated with DNA methyltransferase inhibitors to investigate secondary metabolite contents and expression of related genes and to verify whether DNA methylation is important for regulating secondary metabolite biosynthesis. Common DNA methyltransferase inhibitors used for plant studies are 5-azacytidine (5-azaC), 5-aza-2′-deoxycytidine (5-aza-dC), and zebularine, of which 5-azaC is still the most widely used. In terms of histone deacetylation, HDAC inhibitors are divided into four categories on the basis of structural features. Hydroxamic acid salts, such as trichostatin A (TSA) and diallyl disulfide, are natural HDAC inhibitors50,51. Fatty acids, such as sodium butyrate, competitively bind to the zinc region of RPD3 and HD2-type HDACs to inhibit activity52. Cyclic peptides, including the HC toxin, have a high affinity and specificity for HDACs53,54. Benzamides include sirtinol, which is a specific inhibitor of SIRT1 and SIRT255. Nicotinamide noncompetitively inhibits HDAC activity through sirtuins56. Among these inhibitors, TSA was the first HDAC inhibitor identified, and it is often used in botanical studies. Specifically, TSA enhances histone acetylation and is useful for studying the regulation of gene expression via acetylation modifications. In addition, TSA decreases genomic 5-methylcytosine levels, but the specific mechanism remains unknown57. Thus, DNA methylation and HDAC inhibitor treatments can detect changes in DNA methylation and histone acetylation levels. In addition, changes in related gene expression and secondary metabolite contents can be analyzed to quickly verify the regulatory effects, if any, of DNA methylation and histone acetylation modifications. Regardless, inhibitor treatments cannot elucidate the specific mechanisms underlying DNA methylation and histone acetylation. Moreover, they cannot be employed to identify precise DNA methyltransferases, DNA demethylases, HATs, or HDACs for analysis of the regulatory mechanisms involved. Inhibitor treatments are also relevant for verifying the regulatory effects of DNA methylation and histone acetylation in tea. However, because the genetic background of tea is still unclear and most of the relevant genomic databases are not publicly available, specific DNA methylation and histone acetylation loci in tea cannot be verified by inhibitor treatments. Nevertheless, inhibitor treatment results indicate that DNA methylation and histone acetylation are potentially useful for enhancing the economic value of tea; they may also lead to the development of new strategies for unraveling functional metabolism in tea plants.
According to available studies, there are various techniques for epigenetic research on secondary metabolites, such as genomics, proteomics, transcriptomics, and analysis of specific modification sites58. However, it is difficult to decide which research method to implement. Here, we use DNA methylation and histone acetylation as examples to clarify common experimental techniques for detecting DNA methylation and histone modification levels and discuss their advantages and disadvantages (Table 4).
Many DNA methylation detection techniques have been developed in recent years. These methods can be divided into the following three categories based on the sample DNA preprocessing involved: (1) pretreatment with restriction enzymes59, which mainly includes methylation-sensitive amplification polymorphisms (MSAPs)60 and McrBC61,62; (2) pretreatment with bisulfite treatment63, including high-performance liquid chromatography (HPLC), bisulfite sequencing PCR (BSP)63,64, whole-genome bisulfite sequencing (WGBS)65, third-generation sequencing methods single-molecule real-time (SMRT) sequencing by PacBio66 and Oxford nanopore sequencing67,68; and (3) preconditioning via affinity enrichment69, such as methylated DNA immunoprecipitation (MeDIP) and MeDIP coupled with sequencing (MeDIP-seq)70,71. DNA methylation detection techniques need to be selected according to the specific research objectives, availability of reference genome sequences, and sample size. There are few available methods for studying histone modifications, with ChIP-based methods being the most common72. ChIP-based techniques involve cell fixation, chromatin fragmentation, ChIP, reverse crosslinking, DNA purification, and DNA identification73. ChIP can detect dynamic interaction between trans-factors and DNA in vivo, and it can also be used to study the relationship between various covalent modifications of histones and gene expression. Furthermore, combining ChIP with other methods broadens its utility. For example, ChIP coupled with qPCR (ChIP-qPCR) and ChIP coupled with next-generation short sequence sequencing (ChIP-seq) are currently the most commonly employed methods for detecting histone modifications.
With the combination of biology, physics, and chemistry technology, DNA methylation and histone acetylation detection technologies will continue to improve, and the combination of multiple sequencing technologies can greatly promote the excavation and analysis of epigenetic studies of secondary metabolites in nonmodel plants and accelerate research on the genetic regulation mechanism of DNA methylation and histone acetylation.
Coregulation patterns of DNA methylation and histone acetylation with transcription factors
Many studies have shown that epigenetic regulation is involved in the response to abiotic stress in plants, and this new understanding has resulted in new data and directions for epigenetic research. The epigenetic regulation of secondary metabolism is a complex process, and more studies are needed to explain the mechanism of its establishment and maintenance as well as the relationship between various regulatory factors. Therefore, it is very difficult to study the specific mechanism. The existence of transcription factors greatly solves this problem. Transcription factors generally consist of the following four functional regions: a DNA-binding domain, a transcriptional regulatory region, oligomerization sites, and a nuclear localization signal74. The DNA-binding domain determines the specificity of the binding to cis-elements, whereas the transcriptional regulatory region activates or represses gene expression. A series of transcription factors recently isolated from higher plants were confirmed to regulate the expression of genes responsive to drought, salinity, low temperatures, hormones, pathogens, and wounding. We summarizes the regulatory patterns by which DNA methylation, histone acetylation, and transcription factors may be involved in the coregulation of secondary metabolism in tea, providing methodological references for future studies.
Studies have demonstrated that DNA methylation and histone deacetylation are closely related to the chromatin state, higher-order chromosomal structure, gene transcription, signal transduction, and secondary metabolism21,62. A methylated gene promoter region affects transcription in three ways. Specifically, methylation inhibits the binding of transcriptional activators or promotes that of transcriptional repressors to directly regulate transcription (Fig. 3A)75. In addition, promoting histone modifications that repress transcription (e.g., H3K9me2) and inhibiting histone modifications that enhance transcription (e.g., acetylation) indirectly inhibit transcription (Fig. 3C). Furthermore, methyl-binding proteins (MBDs) bind to methylation sites and interact with other transcriptional repressors to form transcriptional repressor protein complexes (Fig. 3E)76,77,78,79. Histone deacetylation regulates transcription by altering histone acetylation levels of target genes, and they are primarily recruited by and interact with transcription factors (Fig. 3B). The inhibitory effect can be reversed in two ways, resulting in increased histone acetylation levels. First, HDACs disengage from transcription factors targeting specific genes (Fig. 3D). Second, other proteins interact with transcription factors and compete with HDACs to bind to a target gene, thereby decreasing the inhibitory effect of HDACs (Fig. 3F)80,81,82,83.

A DNA methylation inhibits binding by transcription factors. B HDACs recruited by and interact with transcription factors to bind target genes. C Transcriptional regulation of histone modifications (such as H3K9me2 or histone acetylation). D HDACs disengage in interactions with transcription factors to promote the expression of genes. E Methyl-binding proteins (MBDs) bind to methylation sites and interact with other transcriptional repressors to form transcriptional repressor protein complexes to inhibit transcription. F Competitors interact with transcription factors and relieve the inhibitory ability of HDACs. TFs transcription factors, HDACs histone deacetylases, MBDs methyl-binding domain protein.
The specific mechanisms by which epigenetic modifications (i.e., DNA methylation and histone acetylation) and transcription factors coregulate tea secondary metabolism have been investigated based on the regulatory effects of DNA methylation and histone acetylation on tea secondary metabolism as well as the regulatory patterns of DNA methylation and histone acetylation, which revealed that DNA methyltransferases and HDACs have modifying roles (Fig. 2). DNA methyltransferases and HDACs were first identified as modifying enzymes, after which enzyme activity assays and subcellular localization experiments were performed to clarify their functions and regional specificity, providing the basis for research regarding specific regulatory mechanisms. Elucidation of specific regulatory mechanisms as well as the screening and identification of transcription factors are also important. The electrophoretic mobility shift assay (EMSA) can be used to identify transcription factors binding to genes involved in secondary metabolite synthesis. Moreover, transient transcriptional activation assays are useful for examining the regulatory effects of transcription factors on the expression of these genes. DNA methylation and histone acetylation levels of transcription factor genes have also been determined. Differences in histone acetylation levels of transcription factor genes between control and stress conditions imply that histone deacetylation can affect secondary metabolite contents by regulating the histone acetylation of transcription factor genes during secondary metabolism. Differences in the DNA methylation levels of transcription factor genes between control and stress conditions indicate that the methylation status of transcription factor genes affects expression or modulates the binding of upstream transcription factors. If there is no difference in the DNA methylation and histone acetylation levels of transcription factor genes under control and stress conditions, another regulatory mechanism involving other transcription factors and DNA methylation or histone acetylation is likely involved. Furthermore, genes encoding the identified transcription factors, DNA methyltransferases, DNA demethylases, HATs, and HDACs have been cloned, and monoclonal antibodies have been prepared for corresponding in vivo experiments. To further clarify the specific regulatory mechanisms, interactions between transcription factors and DNA methyltransferases, DNA demethylases, HATs, or HDACs have been evaluated by yeast two-hybrid (Y2H), pull-down, bimolecular fluorescence complementation (BiFC), and coimmunoprecipitation (Co-IP) assays. A lack of interactions suggests that transcription are directly regulated by DNA methylation. Methylation of genes mediating secondary metabolite synthesis inhibits transcriptional activators or promotes the binding of transcriptional repressors, which can be analyzed by ChIP-qPCR experiments comparing the binding of transcription factors to secondary metabolite synthesis genes in response to diverse treatments. However, confirmed interactions lead to two possibilities. First, histone acetylation may have important regulatory functions. More specifically, transcription factors recruit HDACs to secondary metabolite synthesis genes and affect the secondary metabolite contents by regulating the acetylation of these genes to control expression levels (Fig. 3B). Second, secondary metabolite synthesis genes are highly methylated. In addition, methyl-binding proteins bind to the DNA sequence and form a transcriptional repressor complex with transcription factors and HATs or HDACs to regulate expression of the secondary metabolite synthesis genes and modulate secondary metabolite contents (Fig. 3E). Finally, transcription factors and DNA methyltransferases, DNA demethylases, HATs, and HDACs can be used in transient transcriptional activation assays to assess their combined effects on the transcriptional regulation of secondary metabolite synthesis genes and to elucidate the underlying mechanisms (Fig. 2). The continual refinement of experimental techniques will allow researchers to conduct transient transcriptional activation assays using tea protoplasts. Compared with transient gene expression in tobacco or Arabidopsis protoplasts, assays involving tea protoplasts eliminate the influence of exogenous genes on the transcription of target genes, resulting in more accurate in vivo data.
It is worth mentioning that there are various modes of epigenetic regulation, and modifying the effects of phosphorylation, ubiquitination, and SUMOylation on epigenetic factors at the protein level has been investigated. For example, in Arabidopsis, siz1-mediated ROS1 SUMOylation enhances the stability of ROS1 and positively regulates active DNA demethylation84. To provide more clarity on research methods for epigenetic and transcription factors in tea plant, the aspects of protein modification of epigenetic factors are not explored here.
Perspectives
Recent epigenetic studies have generated a wealth of new information on DNA methylation and histone acetylation modifications. Moreover, methods for analyzing DNA methylation and histone acetylation have improved, enabling more precise examinations of the regulatory effects of these epigenetic changes on plant gene expression and the synthesis of secondary metabolites.
In the future, several key issues need to be considered or addressed when using these approaches in tea. First, the limitation of materials, which is the key problem, needs to be solved. Due to the lack of a stable genetic transformation system, epigenetic modification research is limited in tea. Therefore, it is urgent to develop an efficient genetic transformation system for tea. Second, the selection of regulatory patterns, from the genomic level to the gene level of specific regulatory mechanisms, is a step-by-step process, and it is necessary to design the most appropriate research strategy for each step. Finally, basic research theory is expected to improve the quality and environmental adaptability of tea.
Compared with Arabidopsis, it is difficult to obtain direct evidence of epigenetic regulation in tea, and the key is the limitation of research materials. In view of the lack of tea plants without corresponding mutants and stable genetic materials, researchers have continuously explored transient expression in tea plants, and virus-induced gene silencing (VIGS)85,86 and antisense oligonucleotide (AsODN) technology comprises a fast and effective method to identify the function of genes in tea and tea epigenetics87,88,89. The latest research reports separation technology of tea protoplasts and localization of biosynthetases of special metabolites, confirming the possibility of transient protoplast expression and providing means for the research on epigenetics in tea90.
The recent development and application of various omics-based techniques (e.g., transcriptomics, genomics, and metabolomics) has resulted in significant breakthroughs in research regarding the evolution of tea species, structural variations, metabolite synthesis, and genetic breeding. Previous studies have clarified the evolution of cultivated tea plants and revealed whole-genome duplication events associated with the genetic diversity among tea resources91,92,93, also elucidating the genetic basis of tea flavors and quality as well as ecological adaptations and providing new insight into the formation and quality of tea flavor-related compounds94,95. Recent studies have also identified molecular and metabolic markers relevant for tea breeding and functional genomics studies96,97. From the perspective of regulatory patterns, the application of multiomics approaches has provided a solid foundation for the study of epigenetic modifications, which will help to clarify the epigenetic regulatory network in tea plants. A more comprehensive analysis may be conducted in conjunction with genomics research methods to identify specific epigenetic regulatory factors and analyze their regulatory mechanism on secondary metabolism in tea plants. It is expected that such research results will be applied for the evaluation and screening of tea plant resources, improvement of tea plant varieties, and targeted regulation of metabolites to enhance the economic value of theoretical research.
Conclusion
DNA methylation and histone acetylation are important modifications that control gene transcription activation and silencing, and they have important regulatory effects on secondary metabolic regulation in tea, but the specific regulatory mechanism remains to be elucidated. Hence, this review proposes a research strategy involving DNA methylation and histone acetylation to regulate secondary metabolism in tea, including rapid identification of DNA methylation and histone acetylation involved in the regulation of secondary metabolism by inhibitor treatment. Changes in the apparent modification level of metabolite synthase genes can be analyzed to identify whether DNA methylation and histone acetylation are involved in the regulation of secondary metabolism. It is also possible to identify whether DNA methylation and histone acetylation are involved in the regulation of secondary metabolism by analyzing changes in expression of DNA methylation- and histone acetylation-related factors under stress. Various possible research modes of DNA methylation and histone acetylation involved in the regulation of tea secondary metabolism have been proposed, and feasible and effective solutions (VIGS, AsODNs, and protoplast transformation) overcome the limitations of the most critical research materials in research strategies. The correct application of this research strategy will promote the development of studies on epigenetic regulation of tea secondary metabolism and reveal key epigenetic information that can be used for future tea genetic breeding.
References
Zeng, L. T., Zhou, X. C., Su, X. G. & Yang, Z. Y. Chinese oolong tea: an aromatic beverage produced under multiple stresses. Trends Food Sci. Tech. 106, 242–253 (2020).
Rawat, R. et al. Characterization of volatile components of Kangra orthodox black tea by gas chromatography-mass spectrometry. Food Chem. 105, 229–235 (2007).
Liao, Y. Y., Zhou, X. C. & Zeng, L. T. How does tea (Camellia sinensis) produce specialized metabolites which determine its unique quality and function: a review. Crit. Rev. Food Sci. Nutr. 1–17 (2021). https://doi.org/10.1080/10408398.2020.1868970.
Zeng, L. T., Watanabe, N. & Yang, Z. Y. Understanding the biosyntheses and stress response mechanisms of aroma compounds in tea (Camellia sinensis) to safely and effectively improve tea aroma. Crit. Rev. Food Sci. Nutr. 59, 2321–2334 (2019).
Yang, Z. Y. et al. Characterisation of volatile and non-volatile metabolites in etiolated leaves of tea (Camellia sinensis) plants in the dark. Food Chem. 135, 2268–2276 (2012).
Yu, Z. M. & Yang, Z. Y. Understanding different regulatory mechanisms of proteinaceous and non-proteinaceous amino acid formation in tea (Camellia sinensis) provides new insights into the safe and effective alteration of tea flavor and function. Crit. Rev. Food Sci. Nutr. 60, 844–858 (2020).
Chen, J. D. et al. The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant. Hortic. Res. 7, 63 (2020).
Zeng, L. T. et al. Formation of volatile tea constituent indole during the oolong tea manufacturing process. J. Agric. Food Chem. 64, 5011–5019 (2016).
Zeng, L. T. et al. Biosynthesis of jasmine lactone in tea (Camellia sinensis) leaves and its formation in response to multiple stresses. J. Agric. Food Chem. 66, 3899–3909 (2018).
Zeng, L. T. et al. Does oolong tea (Camellia sinensis) made from a combination of leaf and stem smell more aromatic than leaf-only tea? Contribution of the stem to oolong tea aroma. Food Chem. 237, 488–498 (2017).
Zhou, Y., Zeng, L. T., Hou, X. L., Liao, Y. Y. & Yang, Z. Y. Low temperature synergistically promotes wounding-induced indole accumulation by INDUCER OF CBF EXPRESSION-mediated alterations of jasmonic acid signaling in Camellia sinensis. J. Exp. Bot. 71, 2172–2185 (2020).
Zhou, Y. et al. Formation of (E)-nerolidol in tea (Camellia sinensis) leaves exposed to multiple stresses during tea manufacturing. Food Chem. 231, 78–86 (2017).
Liu, G. Z. et al. GREEN STRIPE, encoding methylated TOMATO AGAMOUS-LIKE 1, regulates chloroplast development and Chl synthesis in fruit. New Phytol. 228, 302–017 (2020).
Li, W. F. et al. Whole-genome DNA methylation patterns and complex associations with gene expression associated with anthocyanin biosynthesis in apple fruit skin. Planta 250, 1833–1847 (2019).
Ma, C. Q. et al. The effect of promoter methylation on MdMYB1 expression determines the level of anthocyanin accumulation in skins of two non-red apple cultivars. BMC Plant Biol. 18, 108 (2018).
Qian, M. J., Sun, Y. W., Allan, A. C., Teng, Y. W. & Zhang, D. The red sport of ‘Zaosu’ pear and its red-striped pigmentation pattern are associated with demethylation of the PyMYB10 promoter. Phytochemistry 107, 16–23 (2014).
Telias, A. et al. Apple skin patterning is associated with differential expression of MYB10. BMC. Plant Biol. 11, 93 (2011).
Wang, Z. G. et al. The methylation of the PcMYB10 promoter is associated with green-skinned sport in Max Red Bartlett pear. Plant Physiol. 162, 885–896 (2013).
Gu, D. C. et al. Epigenetic regulation of the phytohormone abscisic acid accumulation under dehydration stress during postharvest processing of tea (Camellia sinensis). J. Agric. Food Chem. 69, 1039–1048 (2021).
Yang, J. et al. Involvement of DNA methylation in regulating the accumulation of the aroma compound indole in tea (Camellia sinensis) leaves during postharvest processing. Food Res. Int. 142, 110183 (2021).
Xiao, W. et al. DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 18, 805–814 (2006).
Karan, R., DeLeon, T., Biradar, H. & Subudhi, P. K. Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE 7, e40203 (2012).
Liu, X. C. et al. Transcriptional repression by histone deacetylases in plants. Mol. Plant 7, 764–772 (2014).
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Liu, X. C., Yang, S. G., Yu, C. W., Chen, C. Y. & Wu, K. Q. Histone acetylation and plant development. Enzymes 40, 173–199 (2016).
Ahmad, A., Zhang, Y. & Cao, X. F. Decoding the epigenetic language of plant development. Mol. Plant 3, 719–728 (2010).
Zhao, T., Zhan, Z. P. & Jiang, D. H. Histone modifications and their regulatory roles in plant development and environmental memory. J. Genet. Genomics 46, 467–476 (2019).
Shumakova, O. A., Manyakhin, A. Y. & Kiselev, K. V. Resveratrol content and expression of phenylalanine ammonia-lyase and stilbene synthase genes in cell cultures of Vitis amurensis treated with coumaric acid. Appl. Biochem. Biotechnol. 165, 1427–1436 (2011).
Peng, Z. et al. MiR399d and epigenetic modification comodulate anthocyanin accumulation in Malus leaves suffering from phosphorus deficiency. Plant Cell Environ. 43, 1148–1159 (2019).
Ni, Z. J. et al. Effects of 5-azacytidine on bioactive components of Dendrobium officinale (in Chinese). Zhejiang Agricul. Sci. 7, 1018–1020 (2014).
Fan, D. et al. Histone H3K9 demethylase JMJ25 epigenetically modulates anthocyanin biosynthesis in poplar. Plant J. 96, 1121–1136 (2018).
Quadrana, L. et al. Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 5, 3027 (2014).
Gao, Y. et al. DNA methylation levels in different tissues in tea plant via an optimized HPLC method. Hortic Environ. Biotechnol 60, 967–974 (2019).
Wang, L. et al. DNA methylome analysis provides evidence that the expansion of the tea genome is linked to TE bursts. Plant Biotechnol. J. 17, 826–835 (2019).
Yuan, L. Y., Dai, H. W., Zheng, S. T., Huang, R. & Tong, H. R. Genome-wide identification of the HDAC family proteins and functional characterization of CsHD2C, a HD2-type histone deacetylase gene in tea plant (Camellia sinensis L. O. Kuntze). Plant Physiol. Biochem. 155, 898–913 (2020).
Zhou, Y. H. et al. Changes of DNA methylation levels and patterns in tea plant (Camellia sinensis) during cold acclimation. Acta Agron. Sin. 41, 1047–1055 (2015).
Tong, W. et al. Divergent DNA methylation contributes to duplicated gene evolution and chilling response in tea plants. Plant J. 106, 1312–1327 (2021).
Zhu, C. et al. Genome-wide investigation and transcriptional analysis of cytosine-5 DNA methyltransferase and DNA demethylase gene families in tea plant (Camellia sinensis) under abiotic stress and withering processing. PeerJ 8, e8432 (2020).
Chen, Y. et al. Genome-wide investigation and expression analysis of DNA demethylase genes in tea plant (Camellia sinensis). J. Tea Sci. 41, 28–39 (2021).
Jiang, J. T. et al. Comprehensive proteome analyses of lysine acetylation in tea leaves by sensing nitrogen nutrition. BMC Genomics 19, 1–13 (2018).
Xu, Y. X. et al. Proteome and acetyl-proteome profiling of Camellia sinensis cv.‘Anji Baicha’during periodic albinism reveals alterations in photosynthetic and secondary metabolite biosynthetic pathways. Front. Plant Sci. 8, 2104 (2017).
Zhou, Y. H. et al. Cloning and expression analysis on DNA methyltransferase gene, CsDRM2, of Camellia sinensis. Acta Tea Sin. 56, 1–7 (2015).
Sun, B. M. et al. Purple foliage coloration in tea (Camellia sinensis L.) arises from activation of the R2R3-MYB transcription factor CsAN1. Sci. Rep. 6, 1–15 (2016).
Jing, T. T. et al. UGT85A53 promotes flowering via mediating abscisic acid glucosylation and FLC transcription in Camellia sinensis. J. Exp. Bot. 71, 7018–7029 (2020).
Pastor, V. et al. Fine tuning of reactive oxygen species homeostasis regulates primed immune responses in Arabidopsis. Mol. Plant Microbe Interact. 26, 1334–1344 (2013).
Talbert, P. B. & Henikoff, S. Environmental responses mediated by histone variants. Trends Cell Biol. 24, 642–650 (2014).
Zhu, Y. J. et al. Chromosome-level genome map provides insights into diverse defense mechanisms in the medicinal fungus Ganoderma sinense. Sci. Rep. 5, 11087 (2015).
Di Lorenzo, C., Colombo, F., Biella, S., Stockley, C. & Restani, P. Polyphenols and human Hhealth: the role of bioavailability. Nutrients 13, 273 (2021).
Liao, Y. Y., Fu, X. M., Zeng, L. T. & Yang, Z. Y. Strategies for studying in vivo biochemical formation pathways and multilevel distributions of quality or function-related specialized metabolites in tea (Camellia sinensis). Crit. Rev. Food Sci. Nutr. 11, 1–14 (2020).
Li, H. et al. The histone deacetylase inhibitor trichostatin a promotes totipotency in the male gametophyte. Plant Cell 26, 195–209 (2014).
Wang, Z. et al. Arabidopsis paired amphipathic helix proteins SNL1 and SNL2 redundantly regulate primary seed dormancy via abscisic acid-ethylene antagonism mediated by histone deacetylation. Plant Cell 25, 149–166 (2013).
Chang, S. & Pikaard, C. S. Transcript profiling in Arabidopsis reveals complex responses to global inhibition of DNA methylation and histone deacetylation. J. Biol. Chem. 280, 796–804 (2005).
Murphy, J. P. et al. Histone deacetylase inhibitors and cell proliferation in pea root meristems. Phytochemistry 55, 11–18 (2000).
Darkin-Rattary, S. J. et al. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl Acad. Sci. USA 93, 13143–13147 (1996).
Grozinger, C. M., Chao, E. D., Blackwell, H. E., Moazed, D. & Schreiber, S. L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 276, 38837–38843 (2001).
Bond, D. M., Dennis, E. S., Pogson, B. J. & Finnegan, E. J. Histone acetylation, VERNALIZATION INSENSITIVE 3, FLOWERING LOCUS C, and the vernalization response. Mol. Plant 2, 724–737 (2009).
Law, R. D. & Suttle, J. C. Chromatin remodeling in plant cell culture: patterns of DNA methylation and histone H3 and H4 acetylation vary during growth of asynchronous potato cell suspensions. Plant Physiol. Biochem. 43, 527–534 (2005).
Fang, Y. Y. et al. Research progress and prospect of DNA methylation in perennial woody plants (in Chinese). Sci. Sin. Vitae 50, 154–166 (2020).
Bird, A. P. Use of restriction enzymes to study eukaryotic DNA methylation: II. The symmetry of methylated sites supports semi-conservative copying of the methylation pattern. J. Mol. Biol. 118, 49–60 (1978).
Cervera, M. T., Ruiz-García, L. & Martínez-Zapater, J. M. Analysis of DNA methylation in Arabidopsis thaliana based on methylation-sensitive AFLP markers. Mol. Genet. Genomics 268, 543–552 (2002).
Sutherland, E., Coe, L. & Raleigh, E. A. McrBC: a multisubunit GTP-dependent restriction endonuclease. J. Mol. Biol. 225, 327–348 (1992).
Yang, J. et al. SWI3B and HDA6 interact and are required for transposon silencing in Arabidopsis. Plant J. 102, 809–822 (2020).
Frommer, M. et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA 89, 1827–1831 (1992).
Liu, W. O., Oefner, P. J., Qian, C., Odom, R. S. & Francke, U. Denaturing HPLC-identified novel FBN1 mutations, polymorphisms, and sequence variants in Marfan syndrome and related connective tissue disorders. Genet. Test. 1, 237–242 (1997).
Baubec, T. & Akalin, A. Genome-wide analysis of DNA methylation patterns by high-throughput sequencing. 197–221 (Springer International Publishing, 2016).
Flusberg, B. A. et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465 (2010).
Simpson, J. T. et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–410 (2017).
Rand, A. C. et al. Mapping DNA methylation with high-throughput nanopore sequencing. Nat. Methods 14, 411–413 (2017).
Weber, M. et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 37, 853–862 (2005).
Nair, S. S. et al. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 6, 34–44 (2011).
Xing, X. Y., Zhang, B., Li, D. F. & Wang, T. Comprehensive whole DNA methylome analysis by integrating MeDIP-seq and MRE-seq. Methods Mol. Biol. 1708, 209–246 (2018).
O’Neill, L. P. & Turner, B. M. Immunoprecipitation of chromatin. Method Enzymol. 274, 189–197 (1996).
Nix, D. A., Courdy, S. J. & Boucher, K. M. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinforma. 9, 523 (2008).
Liu, Q., Zhang, G. Y. & Chen, S. Y. Structure and regulation of plant transcription factors. Chin. Sci. Bull. 45, 1465–1474 (2000).
Zhang, H., Lang, Z. & Zhu, J. K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506 (2018).
Curradi, M., Izzo, A., Badaracco, G. & Landsberger, N. Molecular mechanism of gene silencing mediated by DNA methylation. Mol. Cell Biol. 22, 3157–3173 (2002).
Greenberg, M. V. C. & Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol. 20, 590–607 (2019).
Vaucheret, H. et al. Transgene-induced gene silencing in plants. Plant J. 16, 651–659 (1998).
Zhu, H., Wang, G. H. & Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 17, 551–565 (2016).
Gu, D. C. et al. Identification of HDA15-PIF1 as a key repression module directing the transcriptional network of seed germination in the dark. Nucleic Acids Res. 45, 7137–7150 (2017).
Jung, J. H. et al. The cold signaling attenuator HIGH EXPRESSION OF OSMOTICALLY RESPONSIVE GENE1 activates FLOWERING LOCUS C transcription via chromatin remodeling under short-term cold stress in Arabidopsis. Plant Cell 25, 4378–4390 (2013).
Kim, K. C., Lai, Z. B., Fan, B. F. & Chen, Z. X. Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone deacetylase 19 in basal defense. Plant Cell 20, 2357–2371 (2008).
Luo, M. et al. Regulation of flowering time by the histone deacetylase HDA5 in Arabidopsis. Plant J. 8, 925–936 (2015).
Kong, X. F. et al. SIZ1-mediated SUMOylation of ROS1 enhances its stability and positively regulates active DNA demethylation in Arabidopsis. Mol. Plant 13, 1816–1824 (2020).
Burch-Smith, T. M., Andersonm, J. C., Martin, G. B. & Dinesh-Kumar, S. P. Applications and advantages of virus-induced gene silencing for gene function studies in plants. Plant J. 39, 734–746 (2004).
Mondal, T. K., Bhattacharya, A., Ahuja, P. S. & Chand, P. K. Transgenic tea [Camellia sinensis (L.) O. Kuntze cv. Kangra Jat] plants obtained by Agrobacterium-mediated transformation of somatic embryos. Plant Cell Rep. 20, 712–720 (2001).
Chen, Y. X. et al. UGT74AF3 enzymes specifically catalyze the glucosylation of 4-hydroxy-2, 5-dimethylfuran-3(2H)-one, an important volatile compound in Camellia sinensis. Hortic. Res. 7, 25 (2020).
Zhao, M. Y. et al. Sesquiterpene glucosylation mediated by glucosyltransferase UGT91Q2 is involved in the modulation of cold stress tolerance in tea plants. New Phytol. 226, 362–372 (2020).
Liu, G. F. et al. Implementation of CsLIS/NES in linalool biosynthesis involves transcript splicing regulation in Camellia sinensis. Plant Cell Environ 41, 176–186 (2018).
Zhou, Y., Deng, R. F., Xu, X. L. & Yang, Z. Y. Isolation of mesophyll protoplasts from tea (Camellia sinensis) and localization analysis of enzymes involved in the biosynthesis of specialized metabolites. Beverage Plant Res. 1, 2 (2021).
Chen, S. L. et al. (E)-Nerolidol is a volatile signal that induces defenses against insects and pathogens in tea plants. Hortic. Res. 7, 52 (2020).
Wang, X. C. et al. Population sequencing enhances understanding of tea plant evolution. Nat. Commun. 11, 4447 (2020).
Xia, E. H. et al. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol. Plant 13, 1013–1026 (2020).
Xia, E. H. et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant 10, 866–877 (2017).
Wei, C. L. et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl Acad. Sci. USA 115, e4151–e4158 (2018).
Yu, X. M. et al. Metabolite signatures of diverse Camellia sinensis tea populations. Nat. Commun. 11, 5586 (2020).
Zhang, W. Y. et al. Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties. Nat. Commun. 11, 3719 (2020).
Yamamoto, N. et al. Formation of embryogenic cell clumps from carrot epidermal cells is suppressed by 5-azacytidine, a DNA methylation inhibitor. J. Plant Physiol. 162, 47–54 (2005).
Baubec, T., Pecinka, A., Rozhon, W. & Scheid, O. M. Effective, homogeneous and transient interference with cytosine methylation in plant genomic DNA by zebularine. Plant J. 57, 542–554 (2009).
Marfil, C. F., Asurmendi, S. & Masuelli, R. W. Changes in microRNA expression in a wild tuber-bearing Solanum species induced by 5-Azacytidine treatment. Plant Cell Rep. 31, 1449–1461 (2012).
Tyč, D., Nocarová, E., Sikorová, L. & Fischer, L. 5-Azacytidine mediated reactivation of silenced transgenes in potato (Solanum tuberosum) at the whole plant level. Plant Cell Rep. 36, 1311–1322 (2017).
Chen, Z. J. & Pikaard, C. S. Transcriptional analysis of nucleolar dominance in polyploid plants: biased expression/silencing of progenitor rRNA genes is developmentally regulated in Brassica. Proc. Natl Acad. Sci. USA 94, 3442–3447 (1997).
Acknowledgements
Some of the research performed by the authors is supported by financial support from the National Natural Science Foundation of China (31870684, 31902073, and 31922077), the Basic Frontier Science Research Program of the Chinese Academy of Sciences (ZDBS-LY-SM032), the Guangdong Basic and Applied Basic Research Foundation (2020A1515010007), the Regional Key Project of Science and Technology Service Network Plan of Chinese Academy of Sciences (KFJ-STS-QYZX-093), the National Key Research and Development Program of China (2018YFD1000601), the Hangzhou Qianjiang Special Experts Project, the Guangdong Provincial Special Fund For Modern Agriculture Industry Technology Innovation Teams (2020KJ120), and the Foundation of Science and Technology Program of Guangzhou (201804010097). Because of space limitations, we did not cite all publications in the field; we apologize to all colleagues whose work has not been mentioned.
Author information
Authors and Affiliations
Contributions
Y.Z. constructed the manuscript outline. Y.J., G.D., and Y.Z. wrote the manuscript. W.S., Z.X., C.J., L.Y., and Z.L. helped edit the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, J., Gu, D., Wu, S. et al. Feasible strategies for studying the involvement of DNA methylation and histone acetylation in the stress-induced formation of quality-related metabolites in tea (Camellia sinensis). Hortic Res 8, 253 (2021). https://doi.org/10.1038/s41438-021-00679-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-021-00679-9
