
Abstract
The conserved histone variant H2A.Z is essential for transcriptional regulation; defense responses; and various biological processes in plants, such as growth, development, and flowering. However, little is known about how H2A.Z affects the developmental process and ripening of tomato fruits. Here, we utilized the CRISPR/Cas9 gene-editing system to generate a sl_hta9 sl_hta11 double-mutant, designated sl_h2a.z, and found that these two mutations led to a significant reduction in the fresh weight of tomato fruits. Subsequent messenger RNA (mRNA)-seq results showed that dysfunction of Sl_H2A.Z has profound effects on the reprogramming of genome-wide gene expression at different developmental stages of tomato fruits, indicating a ripening-dependent correlation between Sl_H2A.Z and gene expression regulation in tomato fruits. In addition, the expression of three genes, SlPSY1, SlPDS, and SlVDE, encoding the key enzymes in the biosynthesis pathway of carotenoids, was significantly upregulated in the later ripening stages, which was consistent with the increased contents of carotenoids in sl_h2a.z double-mutant fruits. Overall, our study reveals a role of Sl_H2A.Z in the regulation of carotenoids and provides a resource for the study of Sl_H2A.Z-dependent gene expression regulation. Hence, our results provide a link between epigenetic regulation via histone variants and fruit development, suggesting a conceptual framework to understand how histone variants regulate tomato fruit quality.
Similar content being viewed by others


Introduction
In eukaryotes, the packaging of genomic DNA into chromatin is the main form of regulation of gene transcription1,2,3. As the basic unit of chromatin, a nucleosome is composed of an octamer of histones H2A, H2B, H3, and H4, with 146 base pairs of DNA wrapped around the octamer4. Chromatin is usually defined as transcriptionally active euchromatin and compacted inactive heterochromatin, both of which are mainly established and maintained by histone modifications and histone variants5,6. Eukaryotic genomes contain several histone variants that are primarily found among the members of the histone H3 and H2A families7. Among these variants, histone variant H2A.Z in the H2A family has been thoroughly studied in several organisms8,9,10,11,12. In the genome, H2A.Z is greatly enriched at the transcription start site (TSS) of genes5,10,13, which is important for maintaining the activity of genes, reflecting a significant role in the regulation of transcription3,7,14.
Histone variant H2A.Z has been proposed to have either a promotive or repressive effect on transcription, depending on its localization within genes15. In Arabidopsis, H2A.Z within gene bodies has a strong repressive effect on the transcription of stress-response genes15. H2A.Z within the gene body represses the expression of stress-response genes under noninductive conditions, while H2A.Z deposited at the TSS positively or negatively regulates transcription in rice16. H2A.Z occupancy, rather than its distribution, changes simultaneously with transcriptional changes and regulates active DNA demethylation6. Moreover, H2A.Z influences nucleosome stability and chromatin structure to modulate gene expression17. The genome-wide role of H2A.Z in chromatin structure and gene regulation is well established. H2A.Z also has defined regulatory roles in the expression of specific genes involved in development and adaptation18, which, along with its incorporation into chromatin, affirm that H2A.Z is central to transcriptional regulation during developmental and environmental responses19.
In Arabidopsis, there are three functional genes encoding H2A.Z: HISTONE H2A8 (AtHTA8), AtHTA9, and AtHTA1120. In rice, there are three genes (HTA705, HTA712, and HTA713) encoding H2A.Z12. Arabidopsis hta9-1 hta11-1 double mutants exhibit early flowering and have serrated leaves, and such mutations induce the expression of thousands of genes3; however, the Arabidopsis hta9 hta11 double mutant exhibits a less severe phenotype due to the redundancy of HTA8, and this phenotype is confirmed in nearly null h2a.z triple mutants21. More recently, an Arabidopsis complete h2a.z-2 knockout triple mutant generated by CRISPR/Cas9 displays severe growth defects6, indicating the importance of H2A.Z in the regulating plant growth and development. Notably, because Arabidopsis, a model plant species, lacks multiple horticultural processes and traits, such as the production and ripening of fresh fruits, the roles of H2A.Z in regulating fresh fruits of horticultural crop species remain unknown.
Tomato (Solanum lycopersicum) fruits, which are rich in nutrients and have unique flavor, is a widely used model species for studying the ripening and quality of fleshy fruits. As seed-bearing structures of flowering plants, edible fruits are indispensable for humans. Fruits provide important nutrients, including vitamins, carbohydrates, dietary fiber and multiple active substances, such as carotenoids and flavonoids. Moreover, fruit ripening is the key step for the improvement of fruit quality, which involves various physiological, biochemical and structural changes, accompanied by dramatic alterations in color, pigment contents, and carbohydrate contents22,23.
Various studies have suggested that fruit ripening can be driven by multiple regulators at the epigenomic and transcriptomic levels. It has been shown that fruit ripening is influenced by DNA methylation patterns and levels24,25 and that DNA methylation affects the expression of ripening-related genes26,27, suggesting that the activity of numerous metabolic pathways during ripening is potentially associated with DNA methylation and its subsequent effects on the expression of corresponding genes28. Similar to the role of DNA methylation in regulating fruit ripening, histone acetylation was recently reported to be involved in regulating fruit ripening. Silencing the histone deacetylase-encoding gene SlHDT3 inhibits fruit ripening by suppressing ethylene biosynthesis and carotenoid accumulation29. Histone acetylation and H2A.Z deposition are correlated and occur at many genomic regions in Arabidopsis6. The chromatin-remodeling complex SWR1 is recruited to chromatin by recognizing histone acetylation marks and then depositing H2A.Z to the targeted regions to activate DNA demethylation6. DNA methylation, histone acetylation and H2A.Z are related factors in the epigenome. However, the roles of histone variant H2A.Z in the development and ripening of tomato fruit are still unknown.
Here, we characterize the histone variant Sl_H2A.Z and elucidate the roles of Sl_H2A.Z in the development and ripening of tomato fruits. We found that the tomato genome contains three putative Sl_H2A.Z-encoding genes, referred to as Sl_HTA8, Sl_HTA9, and Sl_HTA11. To investigate the genome-wide effect of Sl_H2A.Z on gene expression during fruit ripening, we performed mRNA-seq of tomato fruits of both WT and sl_h2a.z mutant plants at four different ripening stages. We found that double mutations of Sl_H2A.Z have great effects on the reprogramming of genome-wide gene expression in tomato fruits and that Sl_H2A.Z mutations reduce the fresh weight of tomato fruits and increase the contents of carotenoids. Hence, our study provides a valuable resource for the investigation of genes whose expression is regulated by the histone variant Sl_H2A.Z in horticultural crop species.
Results
Characterization of H2A proteins in S. lycopersicum
There are four H2A variants, named H2A, H2A.X, H2A.W and H2A.Z, and these variants define different genomic features, contributing to the overall genomic organization of Arabidopsis5,30. In this study, we found that the S. lycopersicum genome included 12 genes encoding histone Sl_H2A variants, which were grouped into four classes according to their C-terminal conserved motif: Sl_H2A and Sl_H2A.X, Sl_H2A.W, and Sl_H2A.Z (Fig. 1a, b). Two Sl_HTA genes, Sl_HTA1 (Solyc10g006560) and Sl_HTA2 (Solyc12g005270), encoded SlH2A, and three Sl_HTA genes, Sl_HTA3 (Solyc03g005227), Sl_HTA4 (Solyc09g082710), and Sl_HTA5 (Solyc09g074300), were Sl_H2A.X-encoding genes, which harbor the specific SQEF motif5. Furthermore, Sl_HTA6 (Solyc01g099410), Sl_HTA7 (Solyc09g010400), Sl_HTA10 (Solyc11g073250), and Sl_HTA12 (Solyc11g073260) were identified as encoding Sl_H2A.W (Fig. 1a, b), according to the presence of the SPKK motif in the C-terminal region in Arabidopsis31,32. Three of the twelve Sl_HTA genes, Sl_HTA8 (Solyc09g065755), Sl_HTA9 (Solyc12g006830), and Sl_HTA11 (Solyc06g084090), were classified as Sl_H2A.Z-encoding genes (Fig. 1a), with the shortest C-terminal regions among the H2A variants and different L1-loops7 (Fig. 1b).

a Phylogenetic tree of H2A family proteins from Arabidopsis thaliana (At) and S. lycopersicum (Sl). The phylogenetic tree is generated by Geneious software, according to genetic distance model (Jukes-Cantor). The number of the branch in the phylogenetic tree indicates the chance of amino acid substitution per site. b Alignment of S. lycopersicum (Sl) histone H2A variants. Sl_H2A variants are divided into four classes based on their conserved motifs in their C-terminal tails. Sl_H2A.X and Sl_H2A.W have the specific motifs SQEF and SPKK, respectively. Sl_H2A.Z variants have the shortest C-terminal tails among Sl_H2A variants. The L1 loop of Sl_H2A.Z is different from the other H2A variants7.
Generation of sl_hta9 sl_hta11 double-mutant by the CRISPR/Cas9 gene-editing system
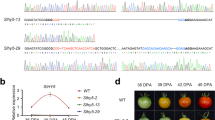
In Arabidopsis, the hta9-1 hta11-1 double-mutant flowers early and has serrated leaves, and the expression of more than two thousand genes is misregulated3. We first found that both Sl_HTA9 and Sl_HTA11 were located in the nucleus (Fig. 2a). To detect whether Sl_H2A.Z regulates tomato fruit ripening, using the CRISPR/Cas9 gene-editing system, we generated a stable loss-of-function sl_hta9 sl_hta11 double-mutant, referred to as sl_h2a.z (Fig. 2b, c)33. We found 8- and 4-bp deletion mutations in sl_hta9-1 and sl_hta11-1, respectively (Fig. 2b, c). We did not separate the homozygous line of the sl_hat9 sl_hta11 sl_hta8 triple mutant, which was probably due to the lethality of the triple mutant, since severe defects in growth and development have already been shown in the complete h2a.z knockout triple mutant in Arabidopsis6.

a The nucleus localization of Sl_HTA9 and Sl_HTA11 in N. benthamiana epidermal cells. b Sequencing results showing mutation in sl_hta9-1 or sl_hta11-1 single mutant generated by CRISPR/Cas9. The genotype in the T2 seedlings of sl_hta9-1 or sl_hta11-1 mutant was confirmed by sequencing. c Schematic diagrams showing the position and nature of mutations in the sl_h2a.z double-mutant. bp, base pair. The sl_h2a.z double-mutant was generated by crossing the sl_hta9-1 single mutant with sl_hta11-1 single mutant. The homozygous sl_h2a.z double-mutant was genotyped by sequencing. d Image of fruits in WT and sl_h2a.z at 34 dpa (MG stage), 40 dpa (B stage), 43 dpa (P stage), and 47 dpa (R stage). dpa, days after pollination. MG, mature green. B, breaker. P, pink. R, red. e The average weight of fresh fruits in WT and sl_h2a.z. *P = 0.019 (n = 30), compared with WT control fruits (2-tailed t-test).
Double mutations of Sl_H2A.Z decreased the fresh weight of tomato fruits
We compared the tomato fruits of the WT and sl_h2a.z double-mutant plants at four stages of tomato fruit ripening: at 34 days after pollination (dpa), referred to as mature green (MG) stage; at 40 dpa, referred to as the breaker (B) stage; at 43 dpa, referred to as the pink (P) stage; and at 47 dpa, referred to as the red (R) stage (Fig. 2d). The dysfunction of Sl_H2A.Z did not visibly change the color of the tomato fruits compared with the WT tomato fruits (Fig. 2d). However, the average weight of fresh fruits significantly decreased in the sl_h2a.z double-mutant fruits compared with the WT fruits (Fig. 2e), suggesting that histone variant H2A.Z regulates the development of tomato fruits. The biomass of 4-week-old seedlings was reduced in the sl_h2a.z double-mutant plants (Fig. S1), which is consistent with the role of H2A.Z in controlling the growth and development of Arabidopsis leaves21,34.
Dysfunction of Sl_H2A.Z and ripening influence the transcript levels of genes in tomato fruits
Incorporation of the histone variant H2A.Z into nucleosomes by the SWR1 chromatin-remodeling complex is a critical step in eukaryotic gene regulation3,15. To elucidate how gene expression is regulated by Sl_H2A.Z during fruit ripening, we carried out a global transcriptome analysis of the tomato fruits at four different fruit ripening stages in both WT and sl_h2a.z plants by mRNA-seq (Fig. 2d). The sequencing quality for all the samples was quite high the raw sequencing data were discarded (Supplemental Tables 1 and 2). The principal component analysis (PCA) data indicated a high consistency among the three biological replicates in each group (Fig. S2a). The Pearson correlation (R2) values between replicates exceeded 95%, indicating high consistency among biological replicates (Fig. S2b). Genes with fragments per kilobase of transcript per million mapped reads (FPKM) values of <1 were defined as barely expressed genes14,35. We divided the expressed genes (FPKM ≥ 1) into three subgroups according to the FPKM percentiles: low expression level (<25th percentile), medium expression level (between the 25th and 75th percentiles), and high expression level (>75th percentile). Interestingly, we found that the numbers of expressed genes (FPKM ≥ 1) gradually decreased during tomato fruit ripening from the MG to R stages, while the numbers of rarely expressed genes gradually increased (Fig. 3a–d). The results presented in the boxplot also confirmed that the transcript levels of genes were decreased (Fig. 3e), suggesting that multiple genes became less inactive with fruit ripening. Double mutations of Sl_H2A.Z did not largely influence the transcript levels of genes in the tomato fruits at any developmental stage, with the exception of the transition from the B to P stage: the trend by which the numbers of expressed genes in the tomato fruits decreased from stage B to stage P was abolished in the sl_h2a.z double-mutant fruits when compared with the WT control fruits (Fig. 3a–d). The heatmap representation of the transcript level showed that both dysfunction of Sl_H2A.Z and ripening obviously reprogrammed genome-wide gene expression patterns (Fig. 3f).

a–d Numbers of genes in the indicated samples at different percentiles of expression level, referred to as 25% high expression (a), medium expression (b), low expression (c), and none expression (d). e Average of expression level of all the annotated genes in S. lycopersicum genome in WT and sl_h2a.z fruits at the four ripening stages. f Heatmap representation of expression level of all the annotated genes in S. lycopersicum genome.
Dysfunction of Sl_H2A.Z induced numerous differentially expressed genes during tomato fruit ripening
Overall, our analysis showed a massive transcriptional reprogramming in response to Sl_H2A.Z mutations (Fig. 3f). We found that dysfunction of Sl_H2A.Z resulted in dysregulation of the expression of multiple genes and novel transcripts in tomato fruits during ripening (Tables S3 and S4). Unlike in WT tomato fruits, the mutations in sl_h2a.z in the fruits of the mutant resulted in 388, 1168, 1098, and 2088 differentially expressed genes (DEGs) whose expression was upregulated at the MG, B, P, and R stages, respectively (Fig. 4a). There were 126, 921, 1143, and 1322 DEGs whose expression was downregulated in the sl_h2a.z tomato fruits at the MG, B, P, and R stages, respectively (Fig. 4a). These results suggest that the expression of numerous genes at different developmental stages of tomato fruits was Sl_H2A.Z-dependent. Moreover, there were 11,814 and 13,458 DEGs in the WT and sl_h2a.z fruits at the R stage compared with the MG stage, respectively (Fig. 4a), and there were 9369 and 8725 DEGs in the WT and sl_h2a.z fruits at the R stage compared with the B stage, respectively (Fig. 4a). There were 7084 and 6975 DEGs in the WT and sl_h2a.z fruits at the R stage compared with the P stage, respectively (Fig. 4a). In the WT fruits, the number of DEGs between the R stage and MG stage was higher than the number between the R stage and B (or P) stage, indicating that gene expression was largely reprogrammed in the process of tomato fruit ripening. Notably, 2553 DEGs were identified in the comparison between the B stage and P stage in the sl_h2a.z fruits, which was much lower than that in the WT fruits (7001 DEGs) (Fig. 4a), suggesting Sl_H2A.Z is essential in the developmental transition of tomato fruits from the B stage to the P stage. In total, 514, 2089, 2241 and 3410 DEGs were identified because of the dysfunction of Sl_H2A.Z at the MG, B, P, and R fruit ripening stages, respectively, showing gradually increasing numbers of reprogrammed genes with increasing fruit ripening (Fig. 4a). Finally, 36 DEGs were common between the WT and sl_h2a.z fruits at the four fruit ripening stages (Fig. 4b, c and Tables S3 and S4), among which the expression of 7 genes was downregulated (Fig. 4d) and the expression of 24 genes was upregulated (Fig. 4e) at all four fruit ripening stages.

a Numbers of DEGs identified in the comparisons between the indicated samples shown in Fig. 2c. b, d, e Overlap of total DEGs (b), overlap of downregulated DEGs (d), and overlap of upregulated DEGs (e) at four ripening stages in sl_h2a.z double-mutant fruits when compared with WT fruits. c Heatmap representation of expression levels in the commonly shared DEGs (shown in b) at all the four ripening stages in WT and sl_h2a.z fruits.
Sl_H2A.Z reprogrammed the expression patterns of ripening-dependent genes
To understand the regulation of ripening and development in the sl_h2a.z double-mutant fruits, we examined ripening-dependent mRNA transcripts in the WT and sl_h2a.z mutant fruits at the four different ripening stages. There were 6386 and 7086 DEGs whose expression was downregulated in the WT and sl_h2a.z fruits, respectively, and the expression of 5428 and 6372 genes was upregulated in the WT and sl_h2a.z fruits, respectively, at the R stage compared to the MG stage (Fig. S3a, b). We calculated the expression levels of the DEGs identified whose expression was downregulated or upregulated in the WT_R vs. WT_MG comparison across all the samples. We found that the expression level of genes in both the WT and sl_h2a.z fruits showed a decreasing trend during fruit ripening, reflected by a cluster of DEGs whose expression was downregulated (Fig. S3c, d). Moreover, the expression level of genes in both the WT and sl_h2a.z fruits displayed an increasing trend during the fruit ripening process, reflected by a cluster of DEGs whose expression was upregulated (Fig. S3e, f). The dysfunction of Sl_H2A.Z largely caused misregulation of the expression patterns of genes in the mutant tomato fruits compared to the WT fruits at the corresponding ripening stage, as reflected by their heatmap representations (Fig. S3c, e) but not by the average transcript level represented in the boxplots (Fig. S3d, f).
Characterizations of the DEGs regulated by ripening and H2A.Z in tomato fruits
To characterize the effects of fruit ripening on the expression of genes in both WT and sl_h2a.z fruits, we compared the DEGs identified from the comparisons among different ripening stages. In total, 2470 DEGs whose expression was upregulated were common among the WT tomato fruits at the B, P, and R stages compared to the MG stages (Fig. S4a, left panel), and 3834 DEGs whose expression was upregulated were common among the sl_h2a.z double-mutant tomato fruits at the B, P, and R stages compared to the MG stages (Fig. S4a, middle panel). In addition, 2,928 DEGs whose expression was downregulated were common in the WT tomato fruits at the B, P, and R stages compared to the MG stage (Fig. S4b, left panel), and 4141 DEGs whose expression was downregulated were common in the sl_h2a.z fruits at the B, P, and R stages compared to the MG stage (Fig. S4b, middle panel). The common DEGs whose expression was upregulated or downregulated in the B vs. MG, P vs. MG, and R vs. MG comparisons were referred to as ripening-dependent upregulated and downregulated DEGs, and the expression of the genes in these clusters was consistently induced by fruit ripening in both the WT and sl_h2a.z fruits.
Among the 2470 ripening-dependent DEGs in the WT fruits, 2243 DEGs overlapped with those in the sl_h2a.z fruits (Fig. S4a, right panel). Among the 2928 ripening-dependent DEGs whose expression was downregulated in the WT fruits, 2547 DEGs overlapped with those in the sl_h2a.z double mutant (Fig. S4b, right panel). There were 1,591 and 1594 DEGs whose expression was upregulated (Fig. S4a, right panel) and downregulated (Fig. S4b, right panel), respectively, showing specific dependence on the function of Sl_H2A.Z, suggesting that Sl_H2A.Z mutations may enable the expression of multiple genes during tomato fruit ripening.
To test whether the Sl_H2A.Z regulation of gene expression is dependent on different ripening stages, we analyzed the DEGs identified in the WT and sl_h2a.z fruits at different sequential developmental stages (i.e., B vs. MG, P vs. B, and R vs. P). From the MG stage to the B stage, the expression of 4241 and 5705 genes was upregulated in the WT and sl_h2a.z fruits, respectively (Fig. S5a, left panel), and the expression of 4475 and 6026 genes was downregulated, respectively (Fig. S5b, left panel). We found that 3732 DEGs whose expression was upregulated (approximately 87.8% of the number in the WT) and 3,933 DEGs whose expression was downregulated (~87.9% of the number in the WT) overlapped between the WT and sl_h2a.z tomato fruits (Fig. S5a, b, left panels). From the B stage to the P stage, the expression of 3442 and 1087 genes was upregulated in WT plants and sl_h2a.z fruits, respectively, and the expression of 3559 and 1466 genes was downregulated, respectively; 826 DEGs whose expression was upregulated (~24.0% of the number in the WT) and 1,169 DEGs whose expression was downregulated (~32.8% of the number in the WT) overlapped in the WT and sl_h2a.z tomato fruits (Fig. S5a, b, middle panels). From the P stage to the R stage, we found that the expression of 3274 and 3519 genes was upregulated in the WT and sl_h2a.z fruits, respectively,and that of 3810 and 3456 genes was downregulated, respectively; in addition, 1599 DEGs whose expression was upregulated (~48.8% of the number in the WT) and 1956 DEGs whose expression was downregulated (~51.3% of the number in the WT) overlapped between the WT and sl_h2a.z fruits (Fig. S5a, b, right panels).
Determination of functionally enrichment in response to Sl_H2A.Z mutations and ripening
To investigate the function of DEGs induced by ripening and Sl_H2A.Z mutations, the DEGs were subjected to Gene Ontology (GO) functional enrichment classification. Generally, the GO classification results indicated that 62, 27, and 34 pathways corresponding to the biological process, molecular function, and cellular component categories, respectively (Fig. 5a and Table S5), were enriched in the ripening-dependent genes whose expression was upregulated in the WT fruits (Fig. S4a, left panel). We found that 186, 50, and 71 pathways corresponding to the biological process, molecular function, and cellular component categories, respectively (Fig. 5a and Table S5), were enriched in the ripening-dependent genes whose expression was upregulated in the sl_h2a.z fruits (Fig. S4a, middle panel). Accordingly, we found that 125, 44, and 30 GO terms were associated with pathways corresponding to biological processes, molecular functions, and cellular components, respectively (Fig. 5b), and were enriched in the ripening-dependent genes whose expression was downregulated in the WT fruits (Fig. S4b, left panel), and we found that 136, 48, and 51 pathways corresponding to biological processes, molecular functions, and cellular components, respectively (Fig. 5b), were enriched in ripening-dependent genes whose expression was downregulated in the sl_h2a.z fruits (Fig. S4b, middle panel). Additionally, we found that 61 GO terms in the biological process category, 23 GO terms in the molecular function category, and 31 GO terms in the cellular component category were enriched in both the WT- and sl_h2a.z-dependent DEGs whose expression was upregulated (Fig. 5a). For the GO terms corresponding to DEGs whose expression was downregulated, the WT and sl_h2a.z tomato fruits shared 100, 34, and 26 enriched pathways associated with the biological process, molecular function, and cellular component categories, respectively (Fig. 5b).

a The numbers of the GOs identified in the ripening-dependent upregulated DEGs (identified in Supplemental Fig. 4a) in WT fruits and sl_h2a.z fruits. b The numbers of the GOs identified in the ripening-dependent downregulated DEGs (identified in Supplemental Fig. 4b) in WT fruits and sl_h2a.z fruits. c The top 33 enriched GOs corresponding to ripening-dependent upregulated DEGs in sl_h2a.z tomato fruits. d The topo 29 enriched GOs corresponding to ripening-dependent downregulated DEGs in sl_h2a.z tomato fruits. The asterisk indicates the GO specifically enriched in sl_h2a.z fruits, but not in the WT fruits in (c, d).
To identify the specific GO terms affected by the dysfunction of Sl_H2A.Z during tomato fruit ripening, the top 33 GO terms corresponding to ripening-dependent DEGs whose expression was upregulated and 29 GO terms corresponding to ripening-dependent DEGs whose expression was downregulated in the biological process category and that were enriched in sl_h2a.z tomato fruits are displayed in Fig. 5c, d. These enriched pathways were specifically associated with the categories “response to nutrient levels”, “proteasomal UBQ-independent protein catabolic process”, “iron-sulfur cluster assembly”, “cell redox homeostasis”, “reactive oxygen species metabolic process”, “mRNA splicing, via spliceosome”, “response to reactive oxygen species”, “positive transcription regulation by Pol II”, “organic cyclic compound catabolic process”, “protein modification by small protein conjugation”, “nucleotide metabolic process”, “carbohydrate derivative biosynthetic process”, “phosphorylation”, “maturation of LSU-rRNA”, “mitotic cell cycle phase transition”, “regulation of response to stress”, “microtubule-based process”, “regulation of cell cycle”, “translation” and so on (Fig. 5c, d), suggesting that dysfunction of Sl_H2A.Z influenced numerous biological processes involved in tomato fruit ripening.
The total carotenoids increased in the sl_h2a.z tomato fruits
Given that the expression of multiple genes and enriched GO pathways were largely induced by mutations in Sl_H2A.Z, whether Sl_H2A.Z regulates the quality of tomato fruits or not is still unknown. To identify the genes whose expression was simultaneously induced both by ripening and by mutations in Sl_H2A.Z, we identified the DEGs that overlapped between the MG stage and R stage in both the WT and sl_h2a.z fruits. Among the 10,297 DEGs common in the WT_R vs. WT_MG and in the sl_h2a.z_R vs. sl_h2a.z_MG comparisons, 1,680 were expressed only in the sl_h2a.z_R vs. WT_R comparison and not in the sl_h2a.z_MG vs. WT_MG comparison (Fig. 6a), suggesting that the expression of genes in this cluster was regulated simultaneously by ripening and Sl_H2A.Z. Among the 1680 DEGs, we found that the PHYTOENE SYNTHASE 1 (SlPSY1) gene, encoding a key enzyme in the pathway of carotenoid biosynthesis, was abundantly expressed (FPKM > 2000 at the R stage) during the ripening process in both WT and sl_h2a.z fruits (Fig. 6b). The transcript level of SlPSY1 largely increased in the sl_h2a.z fruits at the R stage, did the transcript levels of PHYTOENE DESATURASE 1 (SlPDS1) and VIOLAXANTHIN DE-EPOXIDASE (SlVDE), compared to those in the WT fruits (Fig. 6b). SlPDS1 and SlVDE are the genes encoding key enzymes involved in the carotenoid biosynthesis pathway. Using quantitative PCR (qPCR) assays, we confirmed the presence of several genes involved in the pathway of carotenoid biosynthesis, including SlPSY1, SlPDS1, SlVDE, ZETA-CAROTENE DESATURASE (SlZDS), and ZEAXANTHIN EPOXIDASE (SlZEP) (Fig. 6c), whose expression tended to increase in the sl_h2a.z mutant fruits (Fig. 6d). These results indicated that Sl_H2A.Z could regulate the biosynthesis of carotenoids in tomato fruits. We further measured the carotenoid contents in the WT and sl_h2a.z fruits at the P and R stages. The relative index (R2) from the equation of each tested component was >0.99, and the peak pattern of each standard of the indicated carotenoid was clear (Fig. S6a, b), indicating the relatively high accuracy and reliability of the detection method. We found that different carotenoids constituted different proportions in each tested sample (Fig. S6c). Lycopene, (E/Z)-phytoene, lutein, and β-carotene were the main components, and their contents were much higher than those of the other components in both the WT and sl_h2a.z fruits (Fig. 6e and Table S6). There are >600 carotenoids with natural structural variants, which are divided into lycopene, xanthophyll, and carotene compounds36,37,38. We found that the content of (E/Z)-phytoene dramatically increased at the R stage in both the WT and sl_h2a.z fruits compared to the fruits at the P stage, while dysfunction of Sl_H2A.Z did not obviously promote an increase in the (E/Z)-phytoene content in the fruits at either the P or R stage (Fig. 6e, f); the lycopene content slightly (but not significantly) increased in the sl_h2a.z fruits at both the P and R stages compared to those in the WT fruits (Fig. 6f and Table S6). The results showed that the contents of lutein, neoxanthin, violaxanthin, and neoxanthin decreased in the WT fruits at the R stage compared the P stage (Fig. 6f), suggesting that these kinds of carotenoids were degraded with increased ripening. The contents of α- and β-carotene increased in the sl_h2a.z fruits compared to the WT fruits at the P stage (Fig. 6f). The total content of all the tested carotenoids increased in the sl_h2a.z fruits compared to the WT fruits at both the P and R stages (Fig. 6e), which is consistent with the increased expression of genes encoding key enzymes in the carotenoid biosynthesis pathway (such as SlPSY1, SlPDS1, and SlVDE) in the sl_h2a.z mutant fruits.

a Overlapped DEGs among the indicated comparisons. The DEGs include the upregulated and downregulated DEGs. b The FPKM of the SlPSY1, SlPDS1, and SlVDE in the fruits in WT and sl_h2a.z backgrounds at the MG, B, P, and R stages. The values are the mean of the three biological replicates. c The pathway of carotenoids biosynthesis in plants. The key enzymes encoding genes in the pathway are marked. d qPCR detection of SlPSY1, SlPDS, SlVDE, SlZDS, SlZEP, and SlCRTL in the fruits of WT and sl_h2a.z at the MG, B, P, and R stages. Values are means ± SD of three biological replicates. e The contents of the indicated carotenoids in the fruits of WT and sl_h2a.z at the P and R stages. Values are means ± SD of four biological replicates. f Heatmap presentation of the contents of indicated carotenoids in the fruits of WT and sl_h2a.z at the P and R stages. The heatmap is generated by TBtool software based on the values of the contents of the indicated carotenoids.
Discussion
Sl_H2A.Z regulates the fresh weight of tomato fruits
H2A.Z has been widely known to play essential roles in multiple cellular processes by influencing the structure and environment of chromatin in eukaryotes8,19,34,39. In plants, H2A.Z regulates growth and development6,21, phase transitions40, and the response to temperature changes in the environment18,41,42. In this study, we first characterized members of the H2A family in tomato and identified three H2A.Z-encoding genes: Sl_HTA8, Sl_HTA9 and Sl_HTA11. The results showed that mutations of two Sl_H2A.Z-encoding genes, Sl_HTA9 and Sl_HTA11, influence the fresh weight of tomato fruits and the biomass of seedlings, which is consistent with the severe growth defects of Arabidopsis plants caused by the dysfunction of H2A.Z6,21. These results suggest that the conserved histone variant H2A.Z regulates not only vegetative growth (i.e., development of leaves) but also reproductive growth (i.e., development of fresh tomato fruits and flowering) of plants. Given that fruits and vegetables are important components of the human diet, our study on the functions of Sl_H2A.Z in regulating fresh weight and thereby increasing yield highlights the essential roles of histone variants and/or histone modifications in horticultural crop production28.
Dysfunction of Sl_H2A.Z reprograms genome-wide gene expression
Tomato fruit ripening is a highly coordinated and genetically programmed process that includes initiation, maintenance and reorganization of multiple pathways at the transcriptional level to further drive ethylene signals and ripening-related events26,43,44,45,46. These various responses during fruit ripening are directly regulated by gene expression, which is strongly affected by chromatin structure. Chromatin organization can be altered through the incorporation of histone variant H2A.Z5,8,11,47. In this study, both the WT and sl_h2a.z fruits at four different ripening stages were collected for comparison. The mRNA-seq results suggest that Sl_H2A.Z may function in a wide range of communications during tomato fruit ripening and induce intricate genetic regulatory networks through its interaction with ripening. The DEGs identified in different comparisons based on genotype and at different fruit ripening stages, together with the enriched pathways revealed by GO analysis, reflect the key genes (i.e., those shown in Fig. 3) and coordinated regulatory patterns of different biological processes (i.e., those shown in Fig. 5) during the development and ripening of tomato fruits. These changes and variations between the WT and sl_h2a.z fruits reveal coordinated regulatory interactions between histone variants and the transcripts whose levels dynamically change during fruit ripening and provide valuable resources for future studies on fresh fruit biology driven by histone modifications (variants).
Sl_H2A.Z shows dynamic regulation in tomato fruits
Thus far, we have presented comprehensive transcriptional results associated with ripening of fruits of both WT and sl_h2a.z mutant plants at four different ripening stages, revealing the spatial expression patterns associated with ripening in different genotypes at different ripening stages. Previous genomic studies on aging suggest that the expression of highly expressed and highly spliced genes is more likely to be downregulated with age in Drosophila photoreceptors48. Similar to how aging represses gene expression in Drosophila, the gradually increasing numbers of rarely expressed genes and the gradually decreasing numbers of expressed genes (Fig. 3a–d), as well as the reduced transcript levels during tomato fruit ripening (Fig. 3f), suggest that ripening not only reprograms whole-genome-wide gene expression patterns but also represses transcription. Finally, ripening may inactivate the expression of several subsets of genes that are not necessary for or negatively affect ripening. Moreover, similar trends were also found in the sl_h2a.z mutant fruits during ripening (Fig. 3). Unlike in the WT fruits, dysfunction of Sl_H2A.Z inhibits the changes in the gene expression patterns during ripening from the B stage to the P stage, suggesting that the function of Sl_H2A.Z have display time-dependent dynamic characteristics.
Various functions of the histone variant H2A.Z in plants
In Arabidopsis, H2A.Z-containing nucleosomes provide thermosensory information that is used to coordinate the transcriptome under ambient temperature and accurately sense increasing environmental temperatures18. The transcriptome under warm ambient temperature is dependent upon members of the HSFA1 clade of Arabidopsis HSFs, which cause the rapid and dynamic removal of H2A.Z nucleosomes of target genes42. In Brassica napus, high temperatures inhibit the expression of the FT gene through dynamic regulation of H2A.Z and chromatin structure, thus resulting in delayed flowering49. Thermal stress effects on grain yield of Brachypodium distachyon occur via H2A.Z nucleosomes41. In the present study, we found that the numbers of DEGs identified in the sl_h2a.z mutant constitutively increased from 514 to 3,410 during ripening (from the MG stage to the R stage). These results indicate that interactions between H2A.Z and processes involving ripening may occur during the development of tomato fruits, and Sl_H2A.Z shows a developmental stage-dependent function in regulating gene expression, similar to how H2A.Z is sensitive to changing temperatures18. Various plant species show a rapid acceleration of flowering time in response to climate change, especially when experiencing extreme temperatures50. H2A.Z-containing nucleosomes provide specific characteristics applicable to wrapping of DNA, which indicates that there is a set of epigenetic mechanisms in plants to sense temperature changes through fluctuations in nucleosomes18. Chromatin remodeling and nucleosome assembly play an important role in sensing increases in ambient temperature51,52 to regulate flowering. Hence, these studies indicate that the functions of histone variant H2A.Z during the life cycle of plants, such as sensing changes in temperature, accelerating flowering time, and reprogramming gene expression, in tomato fruits are diverse.
Sl_H2A.Z regulates carotenoid biosynthesis in tomato fruits
In this study, we report that dysfunction of Sl_H2A.Z increases the total content of carotenoids by regulating the expression of several genes involved in the carotenoid biosynthesis pathway. The deposition of lycopene is crucial for the characteristic red pigmentation of ripe tomato fruits, and β-carotene is associated with the change from green to red fruit color53. In our study, lycopene, lutein, and β-carotene, the three main carotenoids, increased in the sl_h2a.z fruits at the P stage. The increase in phytoene in both the WT and sl_h2a.z fruits was much higher than that in lycopene during ripening from the P stage to the R stage (Fig. 6e). This can be explained by the following factors: first, since lycopene is the product of the catalytic reaction of phytoene, it is possible that phytoene was not largely converted to lycopene at the sampling/testing stages; second, even though yellow phytoene is not as predominant as red lycopene, phytoene could still contribute to the pigmentation of tomato fruits. We propose that the increase in the contents of total carotenoids in the sl_h2a.z fruits is mainly induced by the upregulated expression of genes involved in the carotenoid biosynthesis pathway (i.e., SlPSY1). Unlike the content of the other three main carotenoids, the lutein content in the sl_h2a.z mutant fruit no longer increased at the R stage, which can be explained by the fact that the enzymes involved in the synthesis and catabolism of lutein are involved in multiple pathways (Fig. 6c). Fruits and vegetables are rich in antioxidant phytochemicals that prevent the risk of chronic diseases induced by oxidative stress54,55,56. Our results suggest that carotenoids function as antioxidants in tomato fruits and are regulated by the histone variant Sl_H2A.Z. Consistent with the role of carotenoids as antioxidants, the GO pathway “cellular response to oxidative stress” was specifically enriched in the sl_h2a.z fruits (Fig. 5c). Since flavor is an important trait of fruits, it is meaningful to investigate the factors influencing tomato flavor caused by dysfunction of histone variants and/or histone modifications in the future.
Multiple factors associated with Sl_H2A.Z function during tomato fruit ripening
In addition to regulating the transcriptome, H2A.Z reportedly prevents DNA methylation at multiple loci57 and interacts with the DNA demethylase ROS1 to mediate DNA demethylation in Arabidopsis6. In tomato, decreased DNA methylation during fruit ripening is mainly induced by the upregulated expression of SlDML226,27. Mutation of SlDML2 inhibits the ripening of tomato fruits by preventing the DNA demethylation of ripening-related genes. Our results suggest that, unlike the function of SlDML2, the dysfunction of Sl_H2A.Z did not obviously influence tomato fruit ripening, which can be explained by the fact functional redundancy concerning Sl_HTA8 occurs and that H2A.Z directs only a subset of ROS1-targeted loci for DNA demethylation6. As detectors of methylation and demethylation, methyl-binding-domain-containing proteins are essential for the organization of methylation patterns in plants58. The expression of Solyc11g068910.2, encoding a methyl-CpG-binding-domain-containing protein, was downregulated in the sl_h2a.z double-mutant fruits at all four developmental and ripening stages (Fig. 4c), suggesting that MBD proteins may play a role in the histone variant-directed reprogramming of the transcriptome during tomato fruit ripening. The detailed function of MBD-containing proteins in fruit ripening needs to be further investigated in knockout mutants generated by the CRISPR/Cas9 gene-editing system.
In addition to the epigenetic and transcriptional regulators induced by H2A.Z, our results show that pathways related to cell redox and reactive oxygen species (ROS) are enriched in Sl_H2A.Z-dependent DEGs (Fig. 5c). Recent studies suggest that tomato fruit ripening is affected by regulation of redox signaling-mediated NOR transcription factors59,60. The redox reactions that function as regulators of activities of enzymes have been reported to be related to DNA methylation, histone methylation, histone acetylation, and chromatin remodeling61. ROS, important signaling factors induced in response to biotic and abiotic stresses in plants62,63,64, mainly control the cell redox state65,66. The response and metabolism of ROS in the enriched pathways identified in our study indicate that ROS may be involved in the regulation directed by the histone variant Sl_H2A.Z during the fruit ripening. The molecular mechanisms underlying ROS and the cell redox state resulting from the dysfunction of Sl_H2A.Z during fruit ripening may help us understand how the dynamic chromatin environment regulated by H2A.Z results in the production of various signals (i.e., secondary messengers) elicited in response to fruit development and ripening. These mechanisms will provide a key direction for breeding crop plants via epigenetic interference. Notably, our results indicate that protein modifications, including phosphorylation and ubiquitination67, are likely involved in the response to fruit ripening; these modifications are probably regulated by H2A.Z. The detailed functions and molecular mechanisms underlying protein modifications during fruit ripening are of interest and can potentially provide more strategies to improve the quality of fresh fruits.
Experimental procedures
Plant material and growth conditions
Wild-type (WT) seeds of the tomato (S. lycopersicum) inbred line 1479 were provided by the Shanghai Academy of Agricultural Sciences, Shanghai, China. All the tomato plants were grown in a greenhouse at the Shanghai Academy of Agricultural Sciences, Shanghai, China.
Protein analysis
Sequences of A. thaliana histone proteins of the H2A family were obtained from the TAIR website (https://www.arabidopsis.org/)5. Sequences of S. lycopersicum histone proteins of the H2A family were obtained from the Sol Genomics Network website (https://solgenomics.net/). The histone H2A variants of S. lycopersicum and A. thaliana were aligned using Geneious software. Phylogenetic tree analysis of the histone H2As of S. lycopersicum and A. thaliana was also performed by Geneious software.
Generation of sl_h2a.z double-mutant by CRISPR
Two 20-bp sgRNA oligos, 5ʹ-GGAGGGAAAGGGCTAGTAGC-3ʹ and 5ʹ- GGGGAAAACAGCGGCTAATA-3ʹ, targeted to Sl_HTA11 (Solyc06g084090) or Sl_HTA9 (Solyc12g006830), respectively, were cloned into the CRISPR/Cas9 gene-editing vector 18T-SlU6-chim. The cassette, which included chimeric RNA driven by the SlU6 promoter and Cas9 driven by the 35S promoter, was subcloned into a pCAMBIA1300 binary vector33. The constructs were subsequently transformed into tomato inbred line 1479 via Agrobacterium infection of leaf explants. Seeds from T2 plants contained homozygous sl_hta9 or sl_hta11 mutation were genotyped by sequencing. The construct in the T1 seedlings of the sl_hta9-1 or sl_hta11-1 mutant was isolated via selfing, which was confirmed by PCR. Finally, we generated sl_h2a.z double-mutant by crossing the sl_hta9-1 and sl_hta11-1 mutants and genotyped the sl_h2a.z double-mutant by sequencing to obtain the homozygous sl_h2a.z double-mutant.
Detection of fresh fruit weight and measurements of aboveground biomass of seedlings
To measure the fresh weight of tomato fruits of the WT and sl_h2a.z plants, 15 plants of each genotype were selected. At 47 dpa, two fruits from the second panicle per plant were harvested. The average weight of the fresh fruits was then calculated (n = 30). To measure the biomass of the WT and sl_h2a.z plants, aboveground parts of 4-week-old seedlings were harvested and measured. Three biological replicates were included. For each biological replicate, the average weight of the aboveground portion of 12 seedlings was calculated.
Nuclear localization assays
Sl_HTA11 and Sl_HTA9 were amplified via PCR from tomato complementary DNAs (cDNAs) and then subcloned into pCambia1300-35S-N1-YFP constructs, which were confirmed by sequencing. Transient expression and image processing were conducted as previously reported68. The constructs containing Sl_HTA11 and Sl_HTA9 were introduced into Agrobacterium tumefaciens (GV3101) by electroporation, after which the transformed bacteria were transiently expressed in tobacco leaves. After 48 h, the tobacco leaves infiltrated with GV3101 were observed under a Delatavison Personal DV system (Applied Precision) using an Olympus UPLanApo water immersion objective lens (60 ×/1.20 numerical apertures). The filters used for DAPI included an exciter (360/40 nm/nm) and an emitter (457/50 nm/nm); for YFP, an exciter (492/18 nm/nm) and an emitter (535/30 nm/nm).
mRNA-seq assays
Tomato fruits of WT and sl_h2a.z plants were harvested at the MG (34 dpa), B (40 dpa), P (43 dpa), and R (47 dpa) stages. For mRNA-seq, pericarp tissues of tomato fruits (n = 15) at each developmental stage from WT or sl_h2a.z plants were mixed together. The samples were frozen immediately in liquid nitrogen and then stored at −80 °C for further analysis. mRNA-seq was performed by Novogene, China.
Briefly, total RNA was extracted from pericarp tissue via TRIzol (Thermo Fisher) and verified on 1% agarose gels. The RNA purity was checked using a NanoPhotometer® spectrophotometer (IMPLEN, USA), and the RNA integrity was assessed using an RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, USA). A total amount of 1 µg of RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using a NEBNext® UltraTM RNA Library Prep Kit for Illumina (NEB, USA), following the manufacturer’s recommendations. Three biological replicates were included for each treatment (sample). Clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the prepared libraries were sequenced on an Illumina NovaSeq platform, and 150 bp paired-end reads were generated.
Data analysis
Quality control
Raw data (raw reads) in fastq format were first processed through in-house Perl scripts. Clean data (clean reads) were obtained by removing reads containing adapters, reads containing poly-N sequences and reads of low quality from the raw data. At the same time, the Q20 value, Q30 value and GC content of the clean data were calculated. All downstream analyses were based on clean data with high quality.
Reads mapped to the reference genome
An index of the reference genome was constructed using HISAT2 v2.0.5, and paired-end clean reads were aligned to the reference genome (Tomato genome version SL3.0) using HISAT2 v2.0.569. We selected HISAT2 as the mapping tool because it can generate a database of splice junctions based on the gene model annotation file and thus provides mapping results that are better than those of other nonsplice mapping tools.
Novel transcript prediction
The mapped reads of each sample were assembled by StringTie (v1.3.3b)70 by a reference-based approach. StringTie uses a novel network flow algorithm as well as an optional de novo assembly step to assemble and quantitate full-length transcripts representing multiple splice variants for each gene locus.
Quantification of gene expression level
FeatureCounts v1.5.0-p3 was used to count the numbers of reads mapped to each gene71. The FPKM of each gene was then calculated based on the length of the gene and read count mapped to that gene72.
Differential expression analysis
Differential expression analysis was performed using the DESeq2 R package (1.16.1)73. Using a model based on the negative binomial distribution, DESeq2 provides statistical routines for determining differences in expression within digital gene expression data. The resulting P-values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with an adjusted P-value < 0.05 and a | log2(fold change)|>0.0 as revealed by DESeq2 were considered differentially expressed genes.
GO enrichment analysis of differentially expressed genes
Gene ontology (GO) enrichment analysis of differentially expressed genes was performed using an online tool (http://geneontology.org/). GO terms with an adjusted P-value < 0.05 were considered significantly enriched
Detection of carotenoids
Carotenoid contents were detected by Metware (http://www.metware.cn/) based on the AB SCIEX QTRAP 6500 liquid chromatography with tandem mass spectrometry (LC-MS/MS) platform. Four biological replicates of each treatment (group) were assessed. In brief, fresh plant materials were freeze dried and stored at −80 °C until extraction. Afterward, <50 mg of dried plant powder for each sample was extracted with a solution buffer (n-hexane:acetone:ethanol). The extract was vortexed for 20 min at room temperature, and the supernatants were collected after centrifugation. The residue was reextracted by repeating the abovementioned steps. The entire supernatant was collected and then evaporated to dryness under a nitrogen stream and reconstituted in a solution buffer (methanol:MTBE). The solution was ultimately filtered through a 0.22 μm filter for further LC-MS analysis.
qPCR assays
Total RNA was extracted by TRIzol (Invitrogen), and a total of 1 μg of mRNA was converted to cDNA with M-MuLV reverse transcriptase (New England Biolabs). The cDNAs were used as templates for real-time PCR with iQ SYBR Green Supermix (Bio-Rad). qPCR was performed with the primers listed in Supplemental Table 7.
Data availability
The mRNA-seq data generated in this publication have been deposited in the National Center for Biotechnology Information database (PRJNA663979).
References
Li, B., Carey, M. & Workman, J. L. The role of chromatin during transcription. Cell 128, 707–719 (2007).
Lai, W. K. M. & Pugh, B. F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol. 18, 548–562 (2017).
Berriri, S., Gangappa, S. N. & Kumar, S. V. SWR1 chromatin-remodeling complex subunits and H2A.Z have non-overlapping functions in immunity and gene regulation in Arabidopsis. Mol. Plant 9, 1051–1065 (2016).
Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 (1997).
Yelagandula, R. et al. The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in Arabidopsis. Cell 158, 98–109 (2014).
Nie, W. F. et al. Histone acetylation recruits the SWR1 complex to regulate active DNA demethylation in Arabidopsis. Proc. Natl Acad. Sci. USA 116, 16641–16650 (2019).
Lei, B. & Berger, F. H2A variants in Arabidopsis: versatile regulators of genome activity. Plant Commun. 1, 100015 (2020).
Kumar, S. V. H2A.Z at the core of transcriptional regulation in plants. Mol. Plant 11, 1112–1114 (2018).
Greenberg, R. S., Long, H. K., Swigut, T. & Wysocka, J. Single amino acid change underlies distinct roles of H2A.Z subtypes in human syndrome. Cell 178, 1421–1436.e1424 (2019).
Ranjan, A. et al. Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell 154, 1232–1245 (2013).
Papamichos-Chronakis, M., Watanabe, S., Rando, O. J. & Peterson, C. L. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 144, 200–213 (2011).
Zhang, K. et al. Differential deposition of H2A.Z in combination with histone modifications within related genes in Oryza sativa callus and seedling. Plant J. 89, 264–277 (2017).
Li, Z. et al. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell 151, 1608–1616 (2012).
Dai, X. et al. H2A.Z represses gene expression by modulating promoter nucleosome structure and enhancer histone modifications in Arabidopsis. Mol. Plant 10, 1274–1292 (2017).
Sura, W. et al. Dual role of the histone variant H2A.Z in transcriptional regulation of stress-response genes. Plant Cell 29, 791–807 (2017).
Zahraeifard, S. et al. Rice H2A.Z negatively regulates genes responsive to nutrient starvation but promotes expression of key housekeeping genes. J. Exp. Bot. 69, 4907–4919 (2018).
Thakar, A. et al. H2A.Z and H3.3 histone variants affect nucleosome structure: biochemical and biophysical studies. Biochem.-Us 48, 10852–10857 (2009).
Kumar, S. V. & Wigge, P. A. H2A.Z-containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell 140, 136–147 (2010).
Talbert, P. B. & Henikoff, S. Environmental responses mediated by histone variants. Trends Cell Biol. 24, 642–650 (2014).
Yi, H. et al. Constitutive expression exposes functional redundancy between the Arabidopsis histone H2A gene HTA1 and other H2A gene family members. Plant Cell 18, 1575–1589 (2006).
Coleman-Derr, D. & Zilberman, D. Deposition of histone variant H2A.Z within gene bodies regulates responsive genes. PLoS Genet. 8, e1002988 (2012).
Seymour, G. B., Ostergaard, L., Chapman, N. H., Knapp, S. & Martin, C. Fruit development and ripening. Annu. Rev. Plant Biol. 64, 219–241 (2013).
Klee, H. J. & Giovannoni, J. J. Genetics and control of tomato fruit ripening and quality attributes. Annu. Rev. Genet. 45, 41–59 (2011).
Teyssier, E. et al. Tissue dependent variations of DNA methylation and endoreduplication levels during tomato fruit development and ripening. Planta 228, 391–399 (2008).
Hadfield, K., Dandekar, A. & Romani, R. Demethylation o fripening specific genes in tomato fruit. Plant Sci. 92, 13–18 (1993).
Lang, Z. et al. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl Acad. Sci. USA 114, E4511–E4519 (2017).
Liu, R. et al. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc. Natl Acad. Sci. USA 112, 10804–10809 (2015).
Tang, D., Gallusci, P. & Lang, Z. Fruit development and epigenetic modifications. New Phytol. https://doi.org/10.1111/nph.16724 (2020).
Guo, J. E. et al. Silencing of histone deacetylase SlHDT3 delays fruit ripening and suppresses carotenoid accumulation in tomato. Plant Sci. 265, 29–38 (2017).
Deal, R. B., Topp, C. N., McKinney, E. C. & Meagher, R. B. Repression of flowering in Arabidopsis requires activation of FLOWERING LOCUS C expression by the histone variant H2A.Z. Plant Cell 19, 74–83 (2007).
Talbert, P. B. et al. A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin 5, 7 (2012).
Kawashima, T. et al. Diversification of histone H2A variants during plant evolution. Trends Plant Sci. 20, 419–425 (2015).
Mao, Y. et al. Application of the CRISPR-Cas system for efficient genome engineering in plants. Mol. Plant 6, 2008–2011 (2013).
March-Diaz, R. & Reyes, J. C. The beauty of being a variant: H2A.Z and the SWR1 complex in plants. Mol. Plant 2, 565–577 (2009).
Huang, H. et al. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl Acad. Sci. USA 116, 1430–1436 (2019).
Milani, A., Basirnejad, M., Shahbazi, S. & Bolhassani, A. Carotenoids: biochemistry, pharmacology and treatment. Br. J. Pharm. 174, 1290–1324 (2017).
Jomova, K. & Valko, M. Health protective effects of carotenoids and their interactions with other biological antioxidants. Eur. J. Med. Chem. 70, 102–110 (2013).
Rutz, J. K., Borges, C. D., Zambiazi, R. C., da Rosa, C. G. & da Silva, M. M. Elaboration of microparticles of carotenoids from natural and synthetic sources for applications in food. Food Chem. 202, 324–333 (2016).
Giaimo, B. D., Ferrante, F., Herchenrother, A., Hake, S. B. & Borggrefe, T. The histone variant H2A.Z in gene regulation. Epigenetics Chromatin 12, 37 (2019).
Gomez-Zambrano, A. et al. Arabidopsis SWC4 binds DNA and recruits the SWR1 complex to modulate histone H2A.Z deposition at key regulatory genes. Mol. Plant 11, 815–832 (2018).
Boden, S. A., Kavanova, M., Finnegan, E. J. & Wigge, P. A. Thermal stress effects on grain yield in Brachypodium distachyon occur via H2A.Z-nucleosomes. Genome Biol. 14, R65 (2013).
Cortijo, S. et al. Transcriptional regulation of the ambient temperature response by H2A.Z nucleosomes and HSF1 transcription factors in Arabidopsis. Mol. Plant 10, 1258–1273 (2017).
Zhong, S. et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 31, 154–159 (2013).
Zuo, J. et al. Relationships between genome methylation, levels of non-coding RNAs, mRNAs and metabolites in ripening tomato fruit. Plant J. https://doi.org/10.1111/tpj.14778 (2020).
Ioannidi, E. et al. Expression profiling of ascorbic acid-related genes during tomato fruit development and ripening and in response to stress conditions. J. Exp. Bot. 60, 663–678 (2009).
Giovannoni, J. J. Genetic regulation of fruit development and ripening. Plant Cell 16, S170–S180 (2004).
Raisner, R. M. et al. Histone variant H2A.Z marks the 5’ ends of both active and inactive genes in euchromatin. Cell 123, 233–248 (2005).
Hall, H. et al. Transcriptome profiling of aging Drosophila photoreceptors reveals gene expression trends that correlate with visual senescence. BMC Genomics 18, 894 (2017).
Del Olmo, I., Poza-Viejo, L., Pineiro, M., Jarillo, J. A. & Crevillen, P. High ambient temperature leads to reduced FT expression and delayed flowering in Brassica rapa via a mechanism associated with H2A.Z dynamics. Plant J. 100, 343–356 (2019).
Fitter, A. H. & Fitter, R. S. R. Rapid changes in flowering time in British plants. Science 296, 1689–1691 (2002).
Franklin, K. A. Plant chromatin feels the heat. Cell 140, 26–28 (2010).
Deal, R. B. & Henikoff, S. Gene regulation: a chromatin thermostat. Nature 463, 887–888 (2010).
Alexander, L. & Grierson, D. Ethylene biosynthesis and action in tomato: a model for climacteric fruit ripening. J. Exp. Bot. 53, 2039–2055 (2002).
Eggersdorfer, M. & Wyss, A. Carotenoids in human nutrition and health. Arch. Biochem. Biophys. 652, 18–26 (2018).
Fiedor, J. & Burda, K. Potential role of carotenoids as antioxidants in human health and disease. Nutrients 6, 466–488 (2014).
Rao, A. V. & Rao, L. G. Carotenoids and human health. Pharm. Res. 55, 207–216 (2007).
Zilberman, D., Coleman-Derr, D., Ballinger, T. & Henikoff, S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456, 125–129 (2008).
Grimanelli, D. & Ingouff, M. DNA methylation readers in plants. J. Mol. Biol. https://doi.org/10.1016/j.jmb.2019.12.043 (2020).
Jiang, G. et al. Redox regulation of the NOR transcription factor is involved in the regulation of fruit ripening in tomato. Plant Physiol. https://doi.org/10.1104/pp.20.00070 (2020).
Li, Y. et al. MicroTom metabolic network: rewiring tomato metabolic regulatory network throughout the growth cycle. Mol. Plant https://doi.org/10.1016/j.molp.2020.06.005. (2020).
Saravana Kumar, R., M. et al. Redox components: key regulators of epigenetic modifications in plants. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21041419 (2020).
Reczek, C. R. & Chandel, N. S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 33, 8–13 (2015).
Gill, S. S. & Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 48, 909–930 (2010).
Hancock, J. T., Desikan, R. & Neill, S. J. Role of reactive oxygen species in cell signalling pathways. Biochem. Soc. Trans. 29, 345–350 (2001).
Schieber, M. & Chandel, N. S. ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 (2014).
Ray, P. D., Huang, B. W. & Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 24, 981–990 (2012).
Meng, X. et al. Proteome-wide lysine acetylation identification in developing rice (Oryza sativa) seeds and protein co-modification by acetylation, succinylation, ubiquitination, and phosphorylation. Biochim. Biophys. Acta Proteins Proteom. 1866, 451–463 (2018).
Shi, L., Wang, J., Hong, F., Spector, D. L. & Fang, Y. Four amino acids guide the assembly or disassembly of Arabidopsis histone H3.3-containing nucleosomes. Proc. Natl Acad. Sci. USA 108, 10574–10578 (2011).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Garber, M., Grabherr, M. G., Guttman, M. & Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 8, 469–477 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
We thank Dr. Q. Yan for assistance with the nuclear localization assays. This work was supported by the National Key R&D Program of China (2016YFD0101703 and 2017YFD0101902), the Natural Science Foundation of Jiangsu Province (BK20200948) and the National Nature Science Foundation of China (32002046).
Author information
Authors and Affiliations
Contributions
X.Y., W.Z., and W.-F.N. designed the research; X. Y., W.-F.N., X.Z., and H.Z. performed the research; W.-F.N. analyzed and interpreted the data; W.-F.N. and X.Y. wrote the manuscript; and W.-F.N. and Y.Y. contributed to the editing of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, X., Zhang, X., Yang, Y. et al. The histone variant Sl_H2A.Z regulates carotenoid biosynthesis and gene expression during tomato fruit ripening. Hortic Res 8, 85 (2021). https://doi.org/10.1038/s41438-021-00520-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-021-00520-3
This article is cited by
-
Genome-wide association studies reveal novel loci for grain size in two-rowed barley (Hordeum vulgare L.)
Theoretical and Applied Genetics (2024)
-
Histone deacetylase gene SlHDA3 is involved in drought and salt response in tomato
Plant Growth Regulation (2023)
-
An apple (Malus domestica) AP2/ERF transcription factor modulates carotenoid accumulation
Horticulture Research (2021)
