
Abstract
Asparagus setaceus is a popular ornamental plant cultivated in tropical and subtropical regions globally. Here, we constructed a chromosome-scale reference genome of A. setaceus to facilitate the investigation of its genome characteristics and evolution. Using a combination of Nanopore long reads, Illumina short reads, 10× Genomics linked reads, and Hi-C data, we generated a high-quality genome assembly of A. setaceus covering 710.15 Mb, accounting for 98.63% of the estimated genome size. A total of 96.85% of the sequences were anchored to ten superscaffolds corresponding to the ten chromosomes. The genome of A. setaceus was predicted to contain 28,410 genes, 25,649 (90.28%) of which were functionally annotated. A total of 65.59% of the genome was occupied by repetitive sequences, among which long terminal repeats were predominant (42.51% of the whole genome). Evolutionary analysis revealed an estimated divergence time of A. setaceus from its close relative A. officinalis of ~9.66 million years ago, and A. setaceus underwent two rounds of whole-genome duplication. In addition, 762 specific gene families, 96 positively selected genes, and 76 resistance (R) genes were detected and functionally predicted in A. setaceus. These findings provide new knowledge about the characteristics and evolution of the A. setaceus genome, and will facilitate comparative genetic and genomic research on the genus Asparagus.
Similar content being viewed by others


Introduction
Asparagus L. is a monocot genus belonging to Asparagaceae (Asparagales) that comprises >200 species distributed widely in regions with an arid–subarid climate in the Old World1,2,3. This genus includes commercially important vegetable species, most prominently A. officinalis, and some species with great ornamental and/or medicinal value, such as A. setaceus and A. cochinchinensis. There are three subgenera in the Asparagus genus: Asparagus, Myrsiphyllum, and Protasparagus2,4,5. Within the Asparagus subgenus, all species are dioecious, whereas the species in the Myrsiphyllum and Protasparagus subgenera are hermaphroditic2. Although this genus has important commercial value, only the model dioecious plant A. officinalis has been extensively investigated, the including sequencing and assembly of a reference genome6,7,8,9,10,11. Other species, especially hermaphroditic species, are poorly investigated. The absence of a reference genome for hermaphroditic species has limited our understanding of the biology and evolution of the Asparagus genus.
Among hermaphroditic Asparagus species, A. setaceus (synonyms: A. plumosus, Protasparagus plumosus, and P. setaceus) is a scrambling perennial herb with needle-like fascicled cladodes12. It is a very popular ornamental plant because of its attractive traits of extremely feathery, soft leaves, and an elegant posture (Fig. 1). A. setaceus also has multiple uses in traditional oriental medicine13. As a wild relative species of the important vegetable A. officinalis, A. setaceus is resistant to purple spot disease caused by infection with Stemphylium vesicarium14 and rust disease caused by infection with Puccinia asparagi5, which are common pathogens of A. officinalis15. Investigating the mechanisms of agricultural characteristics related to pathogen resistance is potentially valuable for the molecular breeding of A. officinalis. Therefore, A. setaceus has high commercial and medicinal value, and is the subject of scientific research because of its properties. However, research on this species is limited. Only a few studies have explored the base chromosome number (2n = 2x = 20) and karyotype of A. setaceus16 and its genome size (~720 Mb)17, micropropagation12, chloroplast genome18, and phylogenetic relationships with other Asparagus species19,20. Genome sequence analysis can greatly promote molecular and genetic studies on this species, and the A. officinalis–A. setaceus genome pair provides a suitable model for the evolutionary analysis of the Asparagus genus.

Image of an A. setaceus plant
In this study, we de novo assembled the genome of A. setaceus through a combination of sequencing strategies, including the use of Nanopore, Illumina, 10× Genomics, and Hi-C technology. Genome annotation, the characterization of genome features, evolutionary analysis, and resistance gene identification were performed based on the assembled genome sequences. Our results provide a foundation for further genome-wide studies on A. setaceus and will be useful for studying the evolution of the Asparagus genus.
Results
Genome sequencing and assembly
A single plant A. setaceus, which has ten homologous pairs of chromosomes in diploid cells (Fig. 2), was used for genome sequencing. The analysis of the 17-mer frequency revealed high genome heterozygosity of 1.9% (Supplementary Fig. 1).
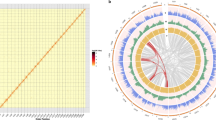
For accurate assembly in this highly heterozygous plant, we sequenced the genome by utilizing a combination of Illumina, Nanopore, 10× Genomics, and Hi-C approaches, and assembled the sequences by using a series of methods. The sequencing and genome assembly workflow is shown in Supplementary Fig. 2a. We obtained a total of 112.52 Gb of Nanopore long reads (Supplementary Fig. 2b) corresponding to ~156.28× coverage of the ~720 Mb A. setaceus genome, as estimated using flow cytometry17. The Nanopore long reads were assembled into contigs by de novo methods. The primary contigs were adjusted with Illumina paired-end reads (84.63 Gb) and then employed for scaffold assembly using 10× Genomics data. The ~180 Gb of 10× Genomics sequencing reads included 2615 contigs grouped into 2061 scaffolds. After redundancy was removed, the assembled genome included 1393 scaffolds with an N50 length of 2.19 Mb. We further connected these scaffolds into superscaffolds by using Hi-C reads (~96 Gb, 133-fold coverage; Supplementary Fig. 2c). Ten of the largest superscaffolds exhibited a total length of 687.77 Mb and matched the ten A. setaceus chromosomes (Supplementary Table 1). The final assembly of the A. setaceus genome was 710.15 Mb in length, constituting 98.63% of the predicted genome size. Among the obtained sequences, 96.85% were anchored to the ten chromosomes (Fig. 3), whereas 22.38 Mb in 657 scaffolds remained unmapped. The sequencing and assembly information are summarized in Table 1.

a Cytological analysis of A. setaceus chromosomes in root tip cells. b Karyotyping of A. setaceus chromosomes based on 45 S rDNA FISH. The 45 S rDNA was labeled with Chroma Tide Alexa Fluor 488 (green), and the chromosomes were counterstained with DAPI (blue). Scale bar, 10 μm

A: Gene density (brown); B: GC content (black); C: density of Gypsy retrotransposable elements (red); D: density of Copia retrotransposable elements (blue); and E: relationship between syntenic blocks, as indicated by lines
Evaluation of the genome assembly
The completeness of this assembled genome was assessed using BUSCO analysis21. Approximately 90.0% of the plant orthologs were included in the assembled sequences (Supplementary Table 2). Furthermore, 89.85% of the transcriptome reads could be mapped to the assembled sequences. In the karyotype of A. setaceus, the first six pairs of chromosomes were clearly larger than the remaining chromosomes (Fig. 2), and the assembly results were consistent with this observation (Fig. 3). These results suggested a high accuracy and completeness of the genome assembly.
Annotation of the A. setaceus genome assembly
We combined different strategies to identify protein-coding genes (Supplementary Fig. 2d). A total of 28,410 genes were identified in the A. setaceus genome (Supplementary Table 3). The average gene length was 6398 bp, and the mean exon number of each gene was 4.95 (Supplementary Table 4). Among these genes, 90.28% showed homology with known genes according to BLAST analysis (Supplementary Table 5). In addition, 2126 noncoding RNAs, including 388 microRNAs (miRNAs), 784 tRNAs, 273 rRNAs, and 681 small nuclear RNAs (snRNAs), were detected (Supplementary Table 6).
Complicated transposable element (TE) annotation showed that 64.43% of the A. setaceus genome assembly was comprised of TEs. Among these TEs, long terminal repeats were predominant, constituting ~42.51% of the assembled genome of A. setaceus (Supplementary Table 7). DNA transposons, long interspersed nuclear elements (LINEs), and short interspersed nuclear elements (SINEs) accounted for 4.12%, 2.90%, and 0.04% of the total assembly, respectively (Supplementary Table 7). Simple sequence repeats (SSRs) are another type of important tandemly repetitive sequences. We used MISA software to detect SSRs in the genome of A. setaceus. A total of 215,955 SSRs (i.e., 85,131 mono-, 103,002 di-, 20,878 tri-, 3683 tetra-, 1967 penta-, and 1294 hexa-nucleotide repeats) were detected (Supplementary Table 8). The total length of the SSR sequences was 8,914,237 bp, accounting for ~1.26% of the assembled A. setaceus genome. Thus, repetitive sequences, including TEs and SSRs, occupied 65.59% of the A. setaceus genome. The basic annotation information is listed in Table 2. The gene density, GC content, Gypsy, and Copia density mapped on the ten A. setaceus chromosomes are shown in a circos plot in Fig. 3.
Evolutionary analysis
Evolutionary analysis was performed by comparing the A. setaceus genome with the genomes of 12 other representative plant species. These species included one other plant in the Asparagus genus (A. officinalis), one additional plant in the Asparagales order (Phalaenopsis equestris), three additional plants in the monocot clade (Oryza sativa, Phoenix dactylifera, and Musa acuminate), and seven other representative plants in the eudicot clade (Amborella trichopoda, Solanum lycopersicum, Arabidopsis thaliana, Carica papaya, Populus trichocarpa, Spinacia oleracea, and Vitis vinifera).
OrthoMCL gene family clustering analysis revealed a total of 13,355 gene families consisting of 21,981 genes in the A. setaceus genome (Fig. 4a, Supplementary Table 9). OrthoMCL clustering recovered 1002 strictly single-copy ortholog gene sets among the 13 analyzed species. Ortholog analysis revealed that A. setaceus, A. officinalis, C. papaya, A. thaliana, and A. trichopoda shared a core set of 7905 gene families (Fig. 4b). Further large-scale analysis among A. setaceus and the 12 other selected species showed that 762 gene families specific to A. setaceus (Supplementary Table 9). These specific genes are mostly involved in heme binding, DNA binding, oxidoreductase activity, and iron and zinc ion binding (Supplementary Fig. 3).

a Clusters of orthologous and paralogous gene families in A. setaceus and 12 other sequenced plant genomes. b Venn diagram representing the distribution of shared gene families among A. setaceus and four other plants (A. officinalis, A. thaliana, C. papaya, and A. trichopoda). c CAFÉ-based estimates of gene family expansions and contractions. The green and red numbers indicate expanded and contracted gene families, respectively. Conserved gene families are indicated in blue in the pi charts. MRCA represents the most recent common ancestor. d Phylogenetic tree of A. setaceus and 12 other species based on a concatenated alignment of 1002 single-copy ortholog gene sets. The tree is rooted with A. trichopoda as the outgroup
Further gene family analysis revealed that 898 gene families were expanded in A. setaceus, whereas 1093 gene families were lost from the A. setaceus genome (Fig. 4c). In comparison with the close relative A. officinalis, which exhibits 558 expanded gene families and 1068 missing gene families, A. setaceus has gained more gene families. These expanded genes present diverse functions, such as binding, protein kinase activity, oxidoreductase activity, and transferase activity. As expected, a phylogenetic analysis showed that A. setaceus displayed a closer relationship to A. officinalis and phylogenetically diverged from the common ancestor ~9.66 million years ago (Mya), after the separation of Orchidaceae at 107.24 Mya (Fig. 4d).
Synteny analysis was performed for the A. setaceus and A. officinalis genomes to understand the genome evolution of these two related species. High collinearity was observed between these two genomes (Fig. 5). The relationships between the chromosomes of A. setaceus and A. officinalis were illustrated based on the shared syntenic blocks. In general, each chromosome of A. setaceus corresponded to one chromosome of A. officinalis. For instance, A. setaceus Chr03 matched the sex chromosome of A. officinalis (Chr01, NC33794.1). In detail, 453 syntenic blocks containing more than three genes were identified from A. setaceus and A. officinalis. The largest synteny block containing 329 genes was found between A. setaceus chromosome 03 and the A. officinalis sex chromosome (01, NC33794.1). Furthermore, the visualization of synteny blocks revealed frequent interchromosomal rearrangement events between the chromosomes of A. setaceus and A. officinalis (Fig. 5). For example, most of the synteny blocks of A. setaceus Chr05 matched A. officinalis Chr05 (NC33798.1), but a few of them corresponded to A. officinalis Chr06 (NC33799.1), Chr07 (NC33800.1), Chr08 (NC33801.1), Chr09 (NC33802.1), or Chr10 (NC33803.1).

Chr01–Chr10 represent the chromosomes of A. setaceus, while NC33794.1–NC33803.1 correspond to the chromosomes of A. officinalis
Genome expansion in A. setaceus
To investigate the genome expansion in A. setaceus, we analyzed whole-genome duplication (WGD) events. 4DTv and Ks values were estimated on the basis of the paralogous gene pairs in collinear regions detected in A. setaceus and three other representative plant species, P. dactylifera, P. equestris, and V. vinifera. The distribution of Ks or 4DTv values in A. setaceus showed two distinct peaks at Ks values of ~0.58 (4DTv ~0.18) and ~1.00 (4DTv ~0.45; Fig. 6a, Supplementary Fig. 4). The first peak was shared by A. setaceus and A. officinalis, and may correspond to the Asparagales-α event previously identified in the A. officinalis genome11. The second is predicted to be derived from a more ancient WGD, which was also found in the A. officinalis genome based on Ks analysis (Supplementary Fig. 4). Dot plots (Fig. 6b) are presented for the paralogs that evolved from the two rounds of WGD events in A. setaceus genome (4–4 diagonal relationships).

a 4DTv distribution in A. setaceus and other representative plant species. b Dot plots of paralogs in the A. setaceus genome, illustrating the two rounds of WGD events
Positively selected genes in A. setaceus
To detect positively selected genes in A. setaceus, we evaluated the Ka/Ks ratios of genes with only one copy by using A. setaceus as a predetermined (foreground) branch and A. officinalis as a background branch. We detected 96 genes that have probably experienced positive selection. GO enrichment revealed that a majority of these genes were involved in ATP binding, nucleic acid binding, oxidoreductase activity, and oxidation–reduction processes (Supplementary Table 10).
Resistance R genes
The A. setaceus genome included 76 resistance (R) genes with nucleotide-binding sites (NBSs). These genes constituted ~0.27% of all A. setaceus genes. Among these genes, 73 resided on the chromosomes, and three genes were located on unmapped scaffolds. These R genes belonged to five groups: TIR-NBS, CC-NBS-LRR, NBS-LRR, NBS, and CC-NBS. NBS-LRR was the largest group, including a total of 29 genes (Supplementary Table 12).
Discussion
A. setaceus is a popular ornamental plant species in many areas of the world. This plant species also has medicinal value. In addition, as a close relative of the important vegetable A. officinalis, comparative genetic and genomic studies on A. setaceus and A. officinalis are helpful for investigating the mechanisms of disease resistance-related agricultural traits, and the origin and evolution of the sex chromosomes of A. officinalis. However, studies on this species are very limited. In particular, molecular-level studies are almost nonexistent. A genome sequence could greatly promote studies on this species and contribute to the comparative analysis of related Asparagus species.
A. setaceus cv. ‘Pyramidalis’ shows a very high level of heterozygosity (1.9%) and a high content of repeats (64.32%). The assembly of such a highly heterozygous and repetitive genome is a challenging task22,23. Thus, we used a series of sequencing strategies, including the Nanopore, Illumina, 10× Genomics, and Hi-C sequencing platforms. We used Nanopore long reads for primary assembly, followed by assembly adjustment with highly accurate short reads. Then, 10× Genomics and Hi-C sequencing data were adopted for scaffold extension and superscaffold (chromosome) construction. By taking full advantage of these sequencing technologies, a chromosome-level genome assembly with high completeness and accuracy was obtained for A. setaceus. BUSCO assessment revealed that 90.0% of the complete BUSCOs could be found in the current assembled A. setaceus genome. This percentage was lower than those in Osmanthus fragrans (96.1%)24 and Brassica oleracea (96.77%)25, but higher than those in the genomes of some other species, such as Ginkgo biloba (73.95%)26 and A. officinalis (88.2%)11. Considering the high level of heterozygosity and repetitiveness of the genome, the current version represents a high-quality genome assembly of A. setaceus.
To guarantee the accuracy of the genome annotation, we integrated various methods to annotate protein-coding genes and used an integrated pipeline to analyze repetitive sequences. A vast majority of the genes in the A. setaceus genome were functionally annotated. Repetitive sequences, mainly consisting of TEs, constitute a major fraction of eukaryotic genomes and play vital roles in genome evolution27,28, chromosome rearrangement29, and gene regulation30. Repetitive sequences occupied 65.59% of the A. setaceus genome assembly; this percentage is very similar to that in the A. officinalis genome (69%).
Comparative genomics analysis showed high synteny and colinearity between the genomes of A. setaceus and A. officinalis. This observation is consistent with their close relationship. These two species diverged from their last common ancestor ~9.66 Mya. Thus, the transition from hermaphroditism to dioecy in Asparagus occurred <9.66 Mya, and the sex chromosome of A. officinalis evolved from the ancestral autosome recently. The results agree with previous findings showing that the cytologically homomorphic X and Y sex chromosomes of A. officinalis are very young19,31,32. However, the accurate timing of the sex chromosome origin will require further analysis. We found that the A. officinalis sex chromosome and A. setaceus Chr03 shared a common ancestral chromosome. Further detailed comparative analyses of these two chromosomes would increase our knowledge of the evolution of the sex chromosome of A. officinalis.
It has been demonstrated that WGD events contribute greatly to the evolution of genomes and genes. Recent evidence has revealed that different plant lineages have experienced distinct WGD events. For example, the grape genome did not experience WGD after the γ-event shared by eudicot plants that took place ~140 Mya, whereas the tea genome underwent two additional rounds of WGD33. The monocot species share a common WGD event, after which different species experienced lineage-specific WGD events34. A previous study revealed that the A. officinalis genome underwent at least two ancient WGDs before the divergence of A. officinalis and other Asparagus species11. The high synteny and collinearity of the genomes of A. setaceus and A. officinalis are in accord with the likely possibility that the two genomes experienced the same WGD events. In this study, the Ks and 4DTv distribution analysis revealed two distinct peaks, which likely correspond to the two rounds of WGD detected in A. officinalis11. The most ancient WGD event was not very clear in the A. officinalis genome based on the current analysis. This may be because of the gene loss process following the WGD events in the A. officinalis genome. The WGD events and subsequent diploidization have contributed greatly to the current genome structure of A. setaceus.
As a wild relative species of garden asparagus, A. setaceus is resistant to some common diseases caused by plant pathogens in A. officinalis5,14,15. In plants, R genes are usually involved in defense mechanisms against infections caused by a majority of specialized plant pathogens35. Thus, examining the resistance genes of A. setaceus is helpful for the further molecular breeding of A. officinalis. Most of the extensively investigated plant R genes contain NBSs35. The A. officinalis genome contains 49 different NBS R genes36. In this study, we identified 76 non-redundant R genes in the A. setaceus genome, which was greater than the number in A. officinalis. Functional studies on these genes would improve our understanding of A. setaceus defense mechanisms and provide a basis for the molecular breeding of A. officinalis.
Conclusion
A chromosome-scale reference genome of A. setaceus (~710.15 Mb) was generated by combining the Nanopore, Illumina, 10× Genomics, and Hi-C sequencing platforms. A total of 28,410 genes were identified. Among these genes, 90.22% were annotated. Repetitive sequences occupied 65.59% of the genome. The divergence between A. setaceus and A. officinalis is estimated to have occurred ~9.66 Mya. Genome evolution analysis provided evidence supporting two rounds of WGD events. The identified genomic features of A. setaceus, including gene families, syntenic blocks, WGD events, and genome-specific genes, provide rich data for comparative genomic studies in plants, especially for studying species in the same genus. The divergence time and synteny analysis between A. setaceus and A. officinalis will contribute to studies on the evolution of the sex chromosome of A. officinalis and the Asparagus genus.
Materials and methods
Molecular karyotype analysis of A. setaceus
A plant of A. setaceus cv. ‘Pyramidalis’ cultivated in the glasshouse of Henan Normal University was used in this study. The preparation of mitotic metaphase spreads, fluorescence in situ hybridization (FISH), and molecular karyotype analysis were performed as previously described37.
Genome sequencing
Total DNA was isolated from young fascicled cladodes and stems by using the CTAB method to construct Nanopore and Illumina libraries. For each Nanopore library, the genomic DNA was fractionated (10–50 kb) with BluePippin (Sage Science, Beverly, MA), repaired, A tailed, adaptor ligated, and used for library construction in accordance with the Nanopore library construction protocol. A total of 67 libraries were generated and sequenced on the GridION X5 sequencer platform (Oxford Nanopore Technologies, UK) at the Nextomics Biosciences Company (Wuhan, China).
10× Genomics linked read sequencing
High-molecular weight DNA extraction, indexing, and barcoding were performed in accordance with the standard protocols provided by 10× Genomics. Approximately 1 ng of sample DNA was used for GEM generation, and 16 bp barcodes were used for the labeling of droplets. After the GEM reactions were thermally amplified, the droplets were fractured, and the intermediate DNA library was purified. Then, the DNA was sheared into 500 bp fragments to construct libraries. Sequencing was performed by using an Illumina HiSeq X Ten sequencer to generate linked reads. The long DNA molecules contained many short reads sharing the same barcode.
Hi-C sequencing
Hi-C sequencing data were generated to obtain physical scaffolds for genome assembly as previously described38. Briefly, fresh spears were harvested, cut into small sections, and immersed in 2% formaldehyde for 15 min for crosslinking. Thereafter, the materials were crushed into a fine powder and used for the isolation of nuclei. The isolated nuclei were purified, digested with Dpn II, blunt-end-repaired, and tagged with biotin. Then, the DNA was religated with the T4 DNA ligation enzyme. After proteinase K digestion and the reversion of formaldehyde crosslinking, biotin-containing DNA fragments were captured and used for the construction of the Hi-C library. The final libraries were sequenced by using an Illumina HiSeq X ten sequencer.
RNA-seq
Total RNA was isolated separately from leaves, stems, and flowers of the same A. setaceus individual by using a QIAGEN RNeasy plant mini kit (QIAGEN, Hilden, Germany). Thereafter, RNA-seq libraries were constructed with a TruSeq RNA library preparation kit (Illumina), and PE150 sequencing was carried out on the HiSeq X ten platform. A total of 7.8 Gb, 7.1 Gb, and 8.5 Gb of sequences were generated from the three sample types. In addition, full-length transcriptome sequencing was conducted for mixed samples by using the PacBio Sequel platform, obtaining an additional 14 Gb of data.
Heterozygosity estimation
The heterozygosity of A. setaceus was estimated via K-mer frequency analysis by using Illumina sequencing data39 in accordance with previously described methods24.
Genome assembly
Oxford Nanopore sequencing data were filtered (mean_qscore > 7) and then employed for genome assembly by using the complete Canu pipeline with default parameters40. The paired-end Illumina reads were mapped to the assembly to improve its accuracy for base-pair correction with BWA MEM41 and Pilon42.
The scaffolds were extended with 10× Genomics data by using ARKS43 with the following parameters: “-c 5 -j 0.55 -m 50-10000 -k 30 -r 0.05 -e 3000 -z 500 -d 0”, and LINKS44 with the following parameters: “-l 5 -a 0.9 -z 500”. Thereafter, redundancy in the assembly was removed using redundans 0-13c45 with the following parameters: “–identity 0.7 –overlap 0.7”.
The Hi-C sequencing data were used for the scaffolding of the preliminary assemblies and to increase the contiguity of the assembly at the chromosome level. The cleaned paired-end reads generated by the Illumina HiSeq platform from the Hi-C library were aligned to the assemblies by using Bowtie2 (version 2.3.2)46. After the map position and orientation of the unique mapped reads were considered, the validated read pairs were filtered. Then, LACHESIS software47, which applies a hierarchical agglomerative clustering strategy, was used for chromosome-level scaffolding by clustering, ordering, and orienting the previous assemblies based on genomic proximity information between Hi-C read pairs. Finally, the adjacent anchored scaffolds were connected using 100 bp Ns to form ten superscaffolds corresponding to ten chromosomes.
Gene and repetitive sequence annotation
Protein-coding genes were identified using strategies that combined de novo gene prediction, experimental evidence obtained from transcriptomic data, and homology-based methods. For homology prediction, GeMoMa48 was used with a protein sequence from A. officinalis, a relative of A. setaceus. For RNA-seq-based prediction, PASA49 was used on the basis of the assembled RNA-seq unigenes. Augustus50 was used for de novo prediction. Then, genes identified by these methods were integrated with EVM51. Then, the sequences of the predicted genes were searched against the commonly used SwissProt, GO, KEGG, KOG, Nr, and InterPro databases for annotation.
For the annotation of noncoding RNAs, tRNAscan-SE software52 was used to predict the tRNAs with eukaryotic parameters. miRNAs, rRNAs, and snRNAs were detected using Infernal cmscan53 to search the Rfam database54. The rRNAs and the corresponding subunits were annotated with RNAmmer v1.255.
Repeat annotation was conducted using RepeatMasker based on a custom library produced using de novo-based and homology-based strategies. The de novo prediction of repeats was carried out by using RepeatModeler. A homology-based detection procedure was performed using a conserved BLASTN search in Repbase56. The consensus families generated by RepeatModeler and repeat sequences with similarity in Repbase were merged as a database to analyze the A. setaceus genome by using RepeatMasker. The genome annotation pipeline is presented in Supplementary Fig. 2d.
Gene family analysis
The protein data of some representative plant species, including A. thaliana, A. setaceus, A. officinalis, A. trichopoda, C. papaya, M. acuminate, O. sativa, P. dactylifera, P. equestris, P. trichocarpa, S. lycopersicum, S. oleracea, and V. vinifera, were retrieved from the NCBI database and used for gene family clustering. All protein sequences were pooled and clustered into different kinds of homologs by using the software OrthoMCL with default parameter settings57.
Phylogenetic tree reconstruction and divergence time prediction
A total of 1002 single-copy genes shared by the analyzed genomes were used for subsequent phylogenetic tree building and divergence time evaluation. The selected protein sequences were concatenated and subjected to multiple alignments by using MAFFT58, and the less regions were filtered using Gblocks59. Then, a phylogenetic tree was constructed using RAxML60, and A. trichopoda was used as the root. The divergence time was estimated using MCMCtree, which was incorporated in the PAML package61. The expansion and contraction of the gene family were analyzed with CAFE (v1.6)62.
Detection of polyploidization events
To detect the polyploidization events in the A. setaceus genome, the protein sequences from A. setaceus were intercompared by using BLASTP (E-value < 1e−05) to identify the conserved paralogs. Protein sequences of P. dactylifera, V. vinifera, and P. equestris were also analyzed, and used for comparison. Then, the WGD events of each species were estimated on the basis of the 4DTv and Ks distributions.
Positively selected gene analysis
To detect the positively selected genes in A. setaceus, the single-copy genes of A. setaceus and the closely related species A. officinalis were aligned using MUSCLE63. Positive selection sites were detected with A. setaceus as a predetermined branch by using Codeml software (part of the PAML program package) with a branch-site model. The positively selected genes were annotated by GO and KEGG analyses.
Identification of resistance (R) genes
To identify R genes, the A. setaceus genome was queried with HMM search by using the HMM profile of the NB-ARC domain (Pfam accession number: PF00931). Then, the NBS domain of the candidate genes was confirmed using the NCBI Conserved Domain Database (CDD)64 and the Pfam database65. The genes without an NBS domain were removed. The confirmed genes belonging to different groups were classified based on the conserved domains that they encoded using the CDD and Pfam databases.
Data availability
Raw data from this study were deposited in the NCBI SRA (Sequence Read Archive) database under the Bioproject ID: PRJNA564485. The genome sequence data (Nanopore, Illumina, 10× Genomics, and Hi-C data) are available under accession numbers SRR10176977, SRR10177257, SRR10176978, SRR10187020, and SRR10187021. Transcriptome data are available under accession numbers SRR10177390, SRR10177391, SRR10186988, and SRR10187001. The assembled genome sequences have been deposited at DDBJ/ENA/GenBank under the accession WHSE00000000. Gene models are available at Dryad (https://doi.org/10.5061/dryad.1c59zw3rm).
References
Dahlgren, R. M. T. et al. (eds) The Families of the Monocotyledons (Springer-Verlag Press, 1985).
Clifford, H. T. & Conran, J. G. Asparagaceae. in Flora of Australia (ed George, A. S.) (Australian Government Publishing Service, 1987).
Kubituki, K. & Rudall, P. J. Asparagaceae. in The Families and Genera of Vascular Plants (ed Kubituki, K.) (Springer-Verlag Press, 1998).
Ellison, J. H. & Kinelski, J. J. Greenwich, a male asparagus hybrid. HortScience 21, 1249 (1986).
Fukuda, T. et al. Molecular phylogeny of the genus Asparagus (Asparagaceae) inferred from plastid petB intron and petD-rpoA intergenic spacer sequences. Plant Spec. Biol. 20, 121–132 (2005).
Li, S. F. et al. Analysis of transposable elements in the genome of Asparagus officinalis from high coverage sequence data. PLoS One 9, e97189 (2014).
Li, S. F. et al. Comparative transcriptome analysis reveals differentially expressed genes associated with sex expression in garden asparagus (Asparagus officinalis). BMC Plant Biol. 17, 143 (2017).
Harkess, A. et al. Sex-biased gene expression in dioecious garden asparagus (Asparagus officinalis). N. Phytol. 207, 883–892 (2015).
Murase, K. et al. MYB transcription factor gene involved in sex determination in Asparagus officinalis. Genes Cells 22, 115–123 (2016).
Dong, T. et al. Anthocyanins accumulation and molecular analysis of correlated genes by metabolome and transcriptome in green and purple asparagus (Asparagus officinalis L.). Food Chem. 271, 18–28 (2019).
Harkess, A. et al. The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nat. Commun. 8, 1279 (2017).
Pindel, A. Regeneration capacity of Asparagus setaceus (Kunth) Jessop ‘Pyramidalis’ in in vitro cultures. Acta Sci. Pol. Hortorum Cultus 16, 85–93 (2017).
McGaw, L. J. & Eloff, J. N. Ethnoveterinary use of southern African plants and scientific evaluation of their medicinal properties. J. Ethnopharmacol. 119, 559–574 (2008).
Bansal, R. K., Menzies, S. A. & Broadhurst, P. G. Screening of Asparagus species for resistance to Stemphylium leaf spot. NZ J. Agric. Res. 29, 539–545 (1986).
Alberti, P. et al. Interspecific hybridization for Asparagus breeding. In Proc of the XLVIII Italian Society of Agricultural Genetics-SIFV-SIGA Joint Meeting (Lecce, Italy, 2004).
Akter, S., Begum, K. N., Sultana, S. S. & Alam, S. S. Karyotype diversity in three Asparagus L. species. Cytologia 82, 551–557 (2017).
Štajner, N., Bohanec, B. & Javornik, B. Genetic variability of economically important Asparagus species as revealed by genome size analysis and rDNA ITS polymorphisms. Plant Sci. 162, 931–937 (2002).
Li, J. R. et al. Characterization of the complete chloroplast genome of Asparagus setaceus. Mitochondrial DNA B 4, 2639–2640 (2019).
Kubota, S., Konno, I. & Kannos, A. Molecular phylogeny of the genus Asparagus (Asparagaceae) explains interspecific crossability between the garden asparagus (A. officinalis) and other Asparagus species. Theor. Appl. Genet. 124, 345–354 (2012).
Norup, M. F. et al. Evolution of Asparagus L. (Asparagaceae): Out-of-South-Africa and multiple origins of sexual dimorphism. Mol. Phylogenet. Evol. 92, 25–44 (2015).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Nowak, M. D. et al. The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biol. 16, 12 (2015).
Schatz, M. C., Witkowski, J. & McCombie, W. R. Current challenges in de novo plant genome sequencing and assembly. Genome Res. 13, 243 (2012).
Yang, X. et al. The chromosome-level quality genome provides insights into the evolution of the biosynthesis genes for aroma compounds of Osmanthus fragrans. Hortic. Res. 5, 72 (2018).
Sun, D. et al. Draft genome sequence of cauliflower (Brassica oleracea L. var. botrytis) provides new insights into the C genome in Brassica species. Hortic. Res. 6, 82 (2019).
Guan, R. et al. Draft genome of the living fossil Ginkgo biloba. Gigascience 5, 49 (2016).
Wicker, T. et al. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 19, 103 (2018).
Samoluk, S. S. et al. Heterochromatin evolution in Arachis investigated through genome-wide analysis of repetitive DNA. Planta 249, 1405–1415 (2019).
Li, S. F. et al. Chromosome evolution in connection with repetitive sequences and epigenetics in plants. Genes 8, 290 (2017).
Uzunović, J., Josephs, E. B., Stinchcombe, J. R. & Wright, S. I. Transposable elements are important contributors to standing variation in gene expression in Capsella Grandiflora. Mol. Biol. Evol. 36, 1734–1745 (2019).
Ming, R., Bendahmane, A. & Renner, S. S. Sex chromosomes in land plants. Annu. Rev. Plant Biol. 62, 485–514 (2011).
Telgmann-Rauber, A., Jamsari, A., Kinney, M. S., Pires, J. C. & Jung, C. Genetic and physical maps around the sex-determining M-locus of the dioecious plant Asparagus. Mol. Genet. Genomics 278, 221–234 (2007).
Wei, C. et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl Acad. Sci. USA 115, E4151–E4158 (2018).
Ming, R. et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 47, 1345–1442 (2015).
Meyers, B. C., Kaushik, S. & Nandety, R. S. Evolving disease resistance genes. Curr. Opin. Plant Biol. 8, 129–134 (2005).
Die, J. V., Castro, P., Millán, T. & Gil, J. Segmental and tandem duplication driving the recent NBS-LRR gene expansion in the asparagus genome. Genes 9, 568 (2018).
Li, S. F. et al. The landscape of transposable elements and satellite DNAs in the genome of a dioecious plant spinach (Spinacia oleracea L.). Mol. DNA 10, 3 (2019).
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting for occurrences K-mers. Bioinformatics 27, 764–760 (2011).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Coombe, L. et al. ARKS: chromosome-scale scaffolding of human genome drafts with linked read kmers. BMC Bioinform. 19, 234 (2018).
Warren, R. L. et al. LINKS: scalable, alignment-free scaffolding of draft genomes with long reads. Gigascience 4, 35 (2015).
Pryszcz, L. P. & Gabaldon, T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44, e113 (2016).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interaction. Nat. Biotechnol. 31, 1119–1125 (2013).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Campbell, M. A., Hass, B. J., Hamilton, J. P., Mount, S. M. & Buell, C. R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7, 327 (2006).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 7, 62 (2006).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mol. DNA 6, 11 (2015).
Li, L. Jr, Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Yang, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556 (1997).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Marchler-Bauer, A. et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226 (2015).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Acknowledgements
This study was funded by the National Natural Science Foundation of China (31970240 and 31770346), the National Key Research and Development Program of China (2016YFD0300203-3), and the Key Scientific Research Projects of Colleges and Universities in Henan Province (19A180003).
Author information
Authors and Affiliations
Contributions
S.-F.L. and W.-J.G. designed the project and wrote the draft manuscript. J.W., R.D., H.-W.Z., and N.L. contributed to the genome evolution analysis, gene family analysis, and resistance gene identification. C.-L.D., Y.-L.Z., and N.L., participated in data analysis and substantively revised the manuscript. The final manuscript has been read and approved by all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, SF., Wang, J., Dong, R. et al. Chromosome-level genome assembly, annotation and evolutionary analysis of the ornamental plant Asparagus setaceus. Hortic Res 7, 48 (2020). https://doi.org/10.1038/s41438-020-0271-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-0271-y
This article is cited by
-
Chromosome-level genome assembly of bunching onion illuminates genome evolution and flavor formation in Allium crops
Nature Communications (2022)
-
Research advances in and prospects of ornamental plant genomics
Horticulture Research (2021)
-
Plant sex chromosomes defy evolutionary models of expanding recombination suppression and genetic degeneration
Nature Plants (2021)
-
The chromosome-level Hemerocallis citrina Borani genome provides new insights into the rutin biosynthesis and the lack of colchicine
Horticulture Research (2021)
-
A chromosome-scale genome assembly of a diploid alfalfa, the progenitor of autotetraploid alfalfa
Horticulture Research (2020)
