
Abstract
Polyploidy is a common phenomenon among willow species. In this study, genome sequencing was conducted for Salix matsudana Koidz (also named Chinese willow), an important greening and arbor tree species, and the genome of this species was compared with those of four other tree species in Salicaceae. The total genome sequence of S. matsudana was 655.72 Mb in size, with repeated sequences accounting for 45.97% of the total length. In total, 531.43 Mb of the genome sequence could be mapped onto 38 chromosomes using the published genetic map as a reference. The genome of S. matsudana could be divided into two groups, the A and B genomes, through homology analysis with the genome of Populus trichocarpa, and the A and B genomes contained 23,985 and 25,107 genes, respectively. 4DTv combined transposon analysis predicted that allotetraploidy in S. matsudana appeared ~4 million years ago. The results from this study will help reveal the evolutionary history of S. matsudana and lay a genetic basis for its breeding.
Similar content being viewed by others
Introduction
Willows, the generic term for tree species in Salix. L and Chosenia Nakai of Salicaceae, include deciduous arbors and shrubs mainly distributed in temperate and frigid zones of the Northern Hemisphere. Willow tree species play important roles in energy production, afforestation, and greening. To date, over 520 willow tree species have been discovered worldwide. Asia is the center of origin for willow, and 257 willow tree species are distributed in China1,2. Willow tree species are resistant to a number of stresses, including salt stress3, drought stress4, water stress5, and heavy metal stress6, and provide important functions in ecological restoration7.
The genetic background of willow is complex. Chromosome ploidy, including diploidy (2n = 12x = 38), triploidy, tetraploidy, pentaploidy, hexaploidy, and even dodecaploidy (2n = 12x = 228), is abundant among willow species in nature8,9. Most shrub willow tree species are diploid, while most arbor species are allopolyploid10,11,12. To provide the necessary genetic characteristics of willow for scientific study, the genomes of several diploid shrub willow species, including Salix suchowensis13 and Salix purpurea (available at http://phytozome.jgi.doe.gov), were sequenced successively.
Salix matsudana Koidz is an allotetraploid arbor tree species in Salix with high stress resistance and strong adaptability. S. matsudana has been cultivated widely in China, and has thus been given the name “Chinese willow”. S. matsudana has been introduced to many areas around the world, including Australia, Europe, and North America14. Selection and breeding of new stress-resistant varieties of S. matsudana have been conducted at several institutions, and a series of elite varieties, including “9901” and “Hailiu 1”, were selected and applied in coastal regions of China. To better understand the molecular characteristics of S. matsudana, the first draft genome of S. matsudana was assembled, and the phylogenetic characteristics of S. matsudana were revealed by comparing its genome with available genomes of S. suchowensis, S. purpurea, Populus trichocarpa15, and Populus euphratica16 in the same family.
Results
Genome assembly and quality assessment
According to the kmer distribution, the heterozygosity rate of S. matsudana was estimated to be 0.71% (Supplementary Fig. S1), demonstrating the highly heterozygous genome of this species. In total, 78.50 G of raw data with a depth of 125× and Q30 of >93.11% was obtained by the second-generation Illumina sequencing platform (Supplementary Table S1); 37.58 G of raw data with a total base content of 35.64 Gb, a mean length of 10.31 Kb, and a mean read quality of 0.834 were obtained by the third-generation PacBio sequencing platform (Supplementary Table S2), and 289 G of optical data were obtained based on standard protocols. The raw data from the third-generation PacBio sequencing platform were initially assembled using Canu17 and Quickmerge18 software, followed by error correction with Pilon19 software using the second-generation sequencing data, and final scaffold extension with Irys-scaffolding20 software using the optical data. Finally, a genome of S. matsudana with a total size of 655.72 Mb was obtained, for which the number of scaffolds, scaffold N50, contig N50, and GC content were 2120, ~12.35 Mb, ~945.75 Kb, and 33.55%, respectively (Table 1).
After quality control of the assembled sequences, 450 core genes accounting for 98.25 of the total (458; Supplementary Table S3) and 1313 complete core genes accounting for 94.38 of the total (1440; Supplementary Table S4) were identified in the CEMGA (Core Eukaryotic Genes Mapping Approach) database and BUSCO database, respectively, demonstrating the acceptable consecutiveness, coverage rate, and accuracy of the S. matsudana genome constructed in this study.
Genome annotation
Analysis of repeated sequences
A repeated sequence database of the S. matsudana genome was constructed based on the principles of structure prediction and de novo sequencing. The genome of S. matsudana was clustered and ranked according to a previously published genetic map21. In total, 531.43 Mb of the genome sequences (accounting for 81.43%) was mapped onto the 38 chromosomes of S. matsudana, the length of which ranged from 8.80–28.42 Mb. In total, 300.41 Mb of repeated sequences, accounting for 45.97% of the whole genome, was identified, among which the numbers of SSRs and unknown repeated sequences were 6185 and 116,059, accounting for 0.51% and 6.76% of the total genome, respectively (Supplementary Table S5).
Gene prediction
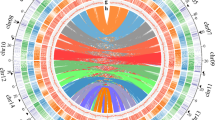
A total of 57,841 genes with a total length of 216,437 Kb, a mean exon length of 229 bp, a mean intron length of 350 bp, and a density of 108.9 genes per Mb were predicted. In addition, 220 miRNAs (belonging to 27 gene families), 321 rRNAs (belonging to 4 gene families), 1557 tRNAs (belonging to 23 gene families), and 3494 pseudogenes were discovered (Fig. 1). The results from blast alignment showed that 99.49% of the genes were annotated in the NR, COG, and KEGG databases (Supplementary Table S6), while only 295 genes were not annotated in these databases.

The outer ring represents the size of individual chromosomes, with a scale of 1 Mb; the second ring (green) represents the distribution of genes; the third ring (red) represents the distribution of repeated sequences; the fourth ring (orange) represents the distribution of pseudogenes; and the fifth ring represents the distribution of tRNAs (orange), miRNAs (green), and rRNAs (red)
Comparisons between genomes of willow and poplar
Comparisons of genome sequences between S. matsudana and four other species in Salicaceae, namely, P. trichocarpa, P. euphratica, S. suchowensis, and S. purpurea, showed that the total number of genes in S. matsudana were 1.53 times and 2.17 times that in S. purpurea and S. suchowensis, respectively (Table 2).
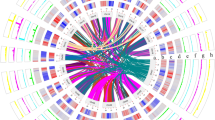
A total of 50,381 of the 57,841 genes in the S. matsudana genome could be clustered into 18,233 gene families, among which 13,787 were common among the five species of Salicaceae, while 302 were specific to S. matsudana (Supplementary Table S7 and Fig. 2). A total of 793 genes were included in the gene families specific to S. matsudana, and their functions were related to metabolic process, cellular process, response to stimulus, and biological regulation (Supplementary Fig. S2).

The numbers under the five tree species represent cluster gene number and total gene family number, respecitively
Phylogenetic analysis of S. matsudana
Expansion analysis of genes in S. matsudana
Further investigation of the expansion and contraction of gene families in the five tree species in Salicaceae demonstrated that the numbers of gene families and genes that were expanded in S. matsudana were 3128 and 20,699, respectively (Supplementary Fig. S3). The functions of these genes were mainly related to metabolic process, cellular process, cell part, cell binding, catalytic activity, and so on. In addition, a series of root development-related gene families, such as root cap, root hair defective 3 GTP-binding protein (RHD3), and transcription factor regulating root and shoot growth via Pin3, were identified as expanded families, and their gene numbers were expanded to 18, 27 and 29, respectively.
Fourfold degenerate site (4DTv) analysis of the five tree species in Salicaceae
The duplication time of the genome was estimated by comparing the 4DTv mutation rates among genes in the collinear fragments of the five tree species in Salicaceae (Fig. 3). A genome-wide replication event was predicted to have occurred 220–358 million years ago (MYA) among the ancestors of Salicaceae, since the first peak was at 0.48 and the synonymous mutation rates (R values) of Salicaceae ranged from 1.09 × 10−9 to 0.67 × 10−9/site/year13. Similarly, a second genome-wide replication event was predicted to have occurred 36.7–59.7 MYA among species in Salicaceae, since the second peak was at 0.08, at which time differentiation between poplar and willow was estimated to have appeared. In addition, the allotetraploid was estimated to have formed 4.6–7.5 MYA in S. matsudana, since a peak appeared at 0.01 for this species.

The x-coordinates and y-coordinates represent 4DTv distance and percentage of syntenic genes, respectively
Long terminal repeat sequence analysis among the five tree species in Salicaceae
Among the long terminal repeat sequence (LTR) transposons in S. matsudana, 213,911, with a total length of 93,513,152 bp and accounting for 14.31% of the repeated sequences, were Copia transposons, while 227,569, with a total length of 100,397,354 bp and accounting for 15.36% of the repeated sequences, were Gypsy transposons.
After estimation of the two types of LTRs by sequence bifurcation analysis, it was found that the insertion of LTR transposons into Salicaceae genomes began 6 MYA, while the insertion of LTR transposons began 4 MYA in the S. matsudana genome (Fig. 4), further confirming that the formation of allotetraploid S. matsudana occurred recently.

The x-coordinates and y-coordinates represent the age of LTRs and number of intact LTR, respectively
Expression analysis of root-related genes in arbor and shrub willows
Arbor and shrub willows are diverse in terms of tiller number, plant height, and primary root development. For instance, the primary roots are well developed in arbor willow, while the primary roots are not obvious, but the lateral roots are well developed in shrub willow. A series of genes were identified as rapidly evolving genes and annotated as root-related genes after comparing the two shrub willow genomes with that of S. matsudana. The expression levels of these genes were determined in ten arbor and shrub willow varieties, using SmACTIN1 as a reference gene. The results showed that TCTP1 and CYP71 were differentially expressed in the roots of arbor and shrub willows (Fig. 5). Results from Arabidopsis showed that TCTP was expressed throughout the plant, but with the highest levels in meristematic regions of the shoot and root, demonstrating its functions in lateral root development, root development, and root hair cell tip growth. CYP71 is a WD40 domain cyclophilin that functions in gene repression, organogenesis, and meristem development. Based on the above results, TCTP1 and CYP71 were predicted to be related to the differentiation of arbor and shrub willows.

The x-coordinates and y-coordinates represent the willow varieties and relative expression, respectively
Analysis of chromosome differentiation and genome comparison in S. matsudana
Chromosome differentiation in the S. matsudana genome
The chromosomes of S. matsudana are displayed in a boxplot according to identity value. Among the 38 chromosomes of S. matsudana, the chromosome group homologous to that of P. trichocarpa was identified and named the A genome, while the remaining 19 chromosomes were classified as the B genome (Supplementary Fig. S4). The A genome was 252.68 Mb in length, with a density of 95 genes per Mb, while the B genome was 278.76 Mb in length, with a density of 90 genes per Mb. The densities of both the A and B genomes of S. matsudana were lower than those in P. trichocarpa.
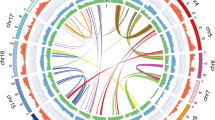
The collinearity between the genomes of S. matsudana and P. trichocarpa was analyzed by MUMMER software. The results showed that the recombination events occurred between Chr1 in P. trichocarpa, and Chr1A and Chr16A in the A genome of S. matsudana, and similar events were also observed for the B genome of S. matsudana (Fig. 6), demonstrating that Chr1 in P. trichocarpa underwent a breakage event. One of the fragments was reconnected with Chr16 and formed Chr1 in S. matsudana, and the other fragment formed Chr1 in S. matsudana, which was consistent with the findings of previous studies22,23.

The x-coordinates and y-coordinates represent the genomes of S. matsudana and P. trichocarpa, respectively. Red represents positive alignment, while blue represents reverse alignment
Comparisons between different genome groups of S. matsudana
Comparisons between different genome groups of S. matsudana showed that the number of genes in the A and B genomes were 23,985 and 25,107, respectively. Collinearity analysis between the A and B genomes revealed different degrees of rearrangement between chromosomes in the A and B genomes, such as between Chr1A and Chr3B, and between Chr1B and Chr3A. In addition, duplications of some chromosome fragments were also observed. For example, Chr8A in the A genome, corresponding to Chr8B in the B genome, was duplicated into Chr10B (Fig. 7).

The x-coordinates and y-coordinates represent the A and B genomes of S. matsudana, respectively
PAVs between the A and B genomes of S. matsudana were initially identified by ppsPCP24 software, further verified by searching for PAV sequences in the reference genome, and finally confirmed by comparing the PAVs with queries. The results indicated that the number of PAV mutation-related genes in the A genome compared with the B genome was 19,841, while the number of PAV mutation-related genes in the B genome compared with the A genome was 12,246. GO enrichment analysis of the genes with PAVs between the A and B genomes suggested that the functions of these genes were consistent in rank between the A and B genomes in terms of biological process, cellular component, and molecular function (Supplementary Table S8 and Supplementary Fig. S5). For example, most of the genes with PAVs, i.e., 3693 in the A genome and 4231 in the B genome, were annotated as being associated with the metabolic process function in the biological process category, accounting for 54.28% and 55.66% of the total genes in the corresponding A and B genomes, respectively.
KEGG enrichment analysis of the genes with PAVs between the A and B genomes suggested that nine of the top ten significantly enriched pathways were different between the A and B genomes. The most enriched pathways were the pyruvate metabolic pathway, and the starch and sucrose metabolic pathway in the A and B genomes, with 40 and 100 genes annotated, respectively (Supplementary Fig. S6). Significant differences between the A and B genomes suggested their varying roles in regulating basal metabolism.
Genome differentiation of willow
A phylogenetic tree was constructed according to the single-copy protein sequences in the five tree species in Salicaceae and the differentiation time of Salicaceae13. The differentiation time of the two shrub willows was ~13.3 MYA, while the differentiation of the original species conferring the A and B genomes to S. matsudana started ~25.2 MYA (Supplementary Fig. S7).
Discussion
Ploidy analysis of willow chromosomes
Willows, the generic term for landscaping and energy tree species in Salicaceae, are variable in terms of chromosome ploidy, from 2n = 38 to 12n = 228 (refs. 8,9). Most shrub willows are diploid, with a genome size of ~400 Mb. For instance, the genome sizes of Salix triandra L. (ref. 25) and S. suchowensis Cheng13,26. are 386 and 425 Mb, respectively. Arbor willows are mainly allopolyploid10,11,12, with a genome size of >650 Mb. For example, the genome sizes of Salix babylonica L. (ref. 27) and Salix alba L. (ref. 25) are 748 and 807 Mb, respectively. S. matsudana is a tetraploid species28. A previous study by our group estimated that the genome size of S. matsudana was ~670 Mb by flow cytometry29. In this study, the genome size of this species was 656 Mb (Table 1), which was in accordance with the size in the previous study.
Chromosome doubling of somatic cells and unreduced germ cells are the main pathways for the formation of polyploids in plants during evolution30. Autopolyploids are polyploids derived from the same species, while allopolyploids are polyploids originating from different species31. For example, hexaploid bread wheat (Triticum aestivum L., AABBDD) originated initially from a hybridization event between Triticum urartu (AA) and T. aestivum L. (BB) to form the tetraploid Triticum turgidum (AABB), followed by the hybridization event between Aegilops tauschii (DD) and T. turgidum32. The origin of tetraploid Gossypium hirsutum L. (AADD) and Gossypium barbadense L. (AADD) was dated back to 1.5 MYA, which involved the formation of the A genome donated by Gossypium herbaceum L. or Gossypium arboreum L., and the hybridization event between the A genome and D genome (donated by Gossypium raimondii Ulbrich)33. The genome comparison in the present study suggested that 19 of the 38 total chromosomes in S. matsudana were consistent with those in the P. trichocarpa genome (Fig. 6), indicating that S. matsudana might have originated from hybridization between the ancestors of P. trichocarpa and another species. 4DTv combined transposon analysis predicted that the genome of S. matsudana doubled ~4 MYA (Figs. 3 and 4), indicating that the formation of the allotetraploid in this species might have occurred during this period.
Split between arbor and shrub willows
Arbors and shrubs are two growth forms specific to tree species. Different types of arbors and shrubs have been observed simultaneously in some willow species34. In this study, the split between the two shrub willow species S. suchowensis and S. purpurea was estimated to have occurred ~13.3 MYA, while the split between the species donating the A genome in S. matsudana and shrub willow occurred ~19.0 MYA (Supplementary Fig. S6). However, because of the limited number of willow genomes sequenced, the exact split time between arbor and shrub willows needs to be further studied.
Arbor and shrub willows displayed significant phenotypic variations in terms of root, branch, and crown growth. The differences in root growth between arbor and shrub willows have been rarely studied, using molecular biology methods. In the current study, TCTP1 and CYP71, screened from rapidly evolving genes of the two shrub willows in comparison to S. matsudana, were identified as differentially expressed genes between the roots of arbor and shrub willow varieties. It was reported that TCTP1 functioned in lateral root development and determined total root length in Arabidopsis35. In addition, TCTP induces the formation of adventitious roots in tobacco36. CYP71 functions in gene repression and organogenesis, and abnormalities in CYP71 result in the ectopic activation of homologous genes regulating meristem development. Mutants of CYP71 displayed phenotypes of low apical meristem activity and disrupted root growth37. TCTP1 and CYP71 were highly expressed in the shrub willow varieties, but weakly expressed in the arbor willow varieties (Fig. 5), suggesting that they might have been involved in the split between arbor and shrub willows. Further functional analysis of these genes would provide insight into the nature of the split between arbor and shrub willows.
Differentiation time between the A and B genomes of S. matsudana
Most species have undergone >2 whole-genome duplications (WGDs), which facilitated their survival and adaptation to different environmental conditions38. The results from this study indicated that species in Salicaceae had undergone at least two WGD events, and the second WGD occurred 36.7–59.7 MYA (Fig. 3).
Fossil records and genome sequences are commonly used to estimate the time of species differentiation. Poplar and willow belong to the same family but different genera. Previous studies based on fossil records revealed that the differentiation of Salicaceae occurred ~45–48 MYA39,40,41. The differentiation time of Salicaceae was estimated to be 12 (ref. 42), 48 (ref. 41), and 52 MYA13 based on sequencing results for the chloroplast, plastid, and nuclear genomes of S. suchowensis, respectively. The variation in differentiation time might be caused by differences in sample selection, which could lead to variation in the correction coefficient and even the final result. The differentiation of the A and B genomes of S. matsudana (Supplementary Fig. S7) was deduced to be 25.2 MYA based on the split time of Salicaceae estimated by the S. suchowensis genome, as well as the single-copy orthologs within the whole genome.
Methods
Genome sequencing
Fresh leaves of S. matsudana were collected in Changjiang town, Rugao city, Jiangsu Province, China, frozen in liquid nitrogen immediately, and stored at −80 °C for subsequent genomic DNA extraction and library construction. The genomic DNA was fragmented by g-TUBE, repaired, ligated with a dumbbell-shaped adaptor, and digested with exonuclease. The target fragments were screened by BluePippin and used for library construction. Second-generation sequencing was performed according to the protocols provided by the Illumina company. Assembly of the genome sequences from the third-generation sequencing was conducted by the PacBio technique. All sequencing procedures were performed by the BioMarker Technologies Company (Beijing, China).
Genome annotation
Annotation of repeated sequences
The repeated sequence database of the S. matsudana genome was constructed initially based on the structure prediction and de novo prediction, using LTR FINDER43, MITE-Hunter44, RepeatScout45, and PILER-DF46 software. The database was classified with PASTEClassifier47 and combined with the Repbase48 database to form the final repeated sequence database. The repeated sequences were annotated based on the final repeated sequence database using RepeatMasker49 software.
Gene expression
Three strategies, namely, de novo prediction, homologous species-based prediction, and unigene-based prediction, were adopted for gene prediction. The prediction results from different strategies were integrated by EVM50 software. GENSCAN51, Augustus52, GlimmerHMM53, GeneID54, and SNAP55 were used for de novo prediction. P. trichocarpa and P. euphratica were selected for homologous species-based prediction using GeMoMa56. PASA57 was used for unigene-based prediction.
Annotation of gene functions
The predicted gene sequences were subjected to BLAST58 alignment with the NR59, COG60, and KEGG61 functional databases. Functional annotations in the GO62 database were compared with the NR database alignment result using Blast2GO63. In addition, the predicted genes were also subjected to COG, KEGG, and GO enrichment analyses.
Comparative genomic analysis
The protein sequences of S. matsudana were aligned with those in related species, including S. suchowensis, S. purpurea, P. trichocarpa, and P. euphratica, to analyze gene duplication within species, gene evolution between species and the classification of species-specific genes. Clustering analysis of gene families was conducted by OrthoMCL64 software. The 4DTv mutation rate was calculated for each of the homologous gene pairs between and within species. LTR sequences with scores ≥6 were detected in the genome by LTR FINDER43 and PS SCAN65, but the repeated sequences detected by LTR FINDER were filtered. The flanking sequences of LTRs were extracted and aligned with MUSCLE66. The degrees of sequence differences were calculated by DistMat software, with the Kimura model and a 7.3 × 10−9 molecular clock.
qRT-PCR of CYP71 and TCTP1
The branches of “2345” (S. suchowensis × S. integra), “51-5” (S. alberti × S. leucopithecia), “2521” (S. purpurea), “52-2” (S. suchowensis × S. leucopithecia), S. integra, S. babylonica, “Yanliu 1” (S. psammophila), “795” (S. matsudana × S. alba), “287” and “Yanjiang” (S. matsudana) were collected and clipped to a length of 10 cm and diameter of 1 cm. Three biological replicates with ten cuttings per replicate were analyzed for each variety. After 15 days of hydroponic culture, the roots of each cutting were collected and stored at −80 °C. Total RNA from the roots of the ten varieties was extracted using an RNAprep Pure Plant kit (Tiangen). The expression levels of the two genes (CYP71 and TCTP1) in each biological replicate were measured by qRT-PCR using three technological replicates. A PrimeScriptTM RT Reagent Kit (Takara) was used for reverse transcription of the RNAs. Reverse transcription was performed at 37 °C for 30 min and 85 °C for 5 s. A One-Step SYBR Primer Script PLUS RT-PCR Kit (Takara) was used for qRT-PCR of the CYP71 and TCTP1 genes. The reaction was performed at 95 °C for 2 min, followed by 40 cycles of 95 °C for 5 s, 56 °C for 30 s, and 72 °C for 20 s. The Actin gene in Salix was used as the reference gene. The qRT-PCR primers are listed in Supplementary Table S9. Relative expression was measured using the 2−ΔΔCt method. The expression level of “287” (S. matsudana) was selected as the reference level (with a relative expression of 1.0).
Construction of the phylogenetic tree
The A and B genomes of S. matsudana, and the genomes of S. suchowensis, S. purpurea, P. trichocarpa, and P. euphratica were used for phylogenetic analysis. The sequence of each single-copy gene family was aligned with MAFFT (v7.205), while the regions with significant differences were removed by Gblocks (v0.91b). Finally, supergenes were obtained by connecting all the aligned gene families in each species. ModelFinder in IQ-TREE was used for model testing, and the best model was predicted to be JTT + F + I + G4. The maximum likelihood method was used to construct a rooted phylogenetic tree. P. trichocarpa was selected as the outgroup species, and the number of bootstraps was set to 1000. Split times and the gradient and Hessian parameters were calculated by MCMCTREE in PAML (v4.9i) software. Visualization of the phylogenetic tree with split times was performed by MCMCTreeR.
Collinearity analysis between the A and B genomes of S. matsudana
The similar gene pairs, with the criteria of an E value < 1e − 5 and a C score > 0.5 (C scores were filtered by JCVI software), were identified by comparing gene sequences between species using Diamond (v0.9.29.130). gff3 was used to determine whether similar gene pairs were adjacent in the genome.
References
Gullberg, U. Towards making willows pilot species for coppicing production. For. Chron. 69, 721–726 (1993).
Lindegaard, K. N. & Barker, J. Breeding willows for biomass. Asp. Appl Biol. 49, 155–162 (1997).
Zhang, J. et al. The genetic architecture of growth traits in Salix matsudana under salt stress. Hortic. Res. 4, 17024 (2017).
Polt, P., Sjödin, P., Weih, M., Rönnberg-Wästljung, A. C. & Berlin, S. Genome-wide transcriptional and physiological responses to drought stress in leaves and roots of two willow genotypes. BMC Plant Biol. 15, 244 (2015).
Hangs, R. D., Schoenau, J. J., Van Rees, K. & Steppuhn, H. Examining the salt tolerance of willow (Salix spp.) bioenergy species for use on salt-affected agricultural lands. Can. J. Plant Sci. 91, 509–517 (2011).
Yanitch, A. et al. Transcriptomic response of purple willow (Salix purpurea) to arsenic stress. Front. Plant Sci. 8, 1115 (2017).
Aronsson, P., Dahlin, T. & Dimitriou, I. Treatment of landfill leachate by irrigation of willow coppice–plant response and treatment efficiency. Environ. Pollut. 158, 795–804 (2010).
Suda, Y. & Argus, G. W. Chromosome numbers of some North American Salix. Brittonia 20, 191–197 (1968).
Macalpine, W. J., Shield, I. F., Trybush, S. O., Hayes, C. M. & Karp, A. Overcoming barriers to crossing in willow (Salix spp.) breeding. Asp. Appl. Biol. 90, 173–180 (2008).
Argus, G. W. Infrageneric classification of Salix (Salicaceae) in the new world. Syst. Bot. Monogr. 52, 1–121 (1997).
Barcaccia, G., Meneghetti, S., Albertini, E., Triest, L. & Lucchin, M. Linkage mapping in tetraploid willows: segregation of molecular markers and estimation of linkage phases support an allotetraploid structure for Salix alba× Salix fragilis interspecific hybrids. Heredity 90, 169–180 (2003).
Barcaccia, G., Meneghetti, S., Lucchin, M. & De Jong, H. Genetic segregation and genomic hybridization patterns support an allotetraploid structure and disomic inheritance for Salix species. Diversity 6, 633–651 (2014).
Dai, X. et al. The willow genome and divergent evolution from poplar after the common genome duplication. Cell Res. 24, 1274–1277 (2014).
Qiao, G. et al. Comparative proteomic analysis of responses to salt stress in Chinese willow (Salix matsudana Koidz). Plant Mol. Biol. Rep. 32, 814–827 (2014).
Tuskan, G. A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604 (2006).
Ma, T. et al. Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 4, 1–9 (2013).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Chakraborty, M. et al. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 44, e147 (2016).
Chin, C. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Jennifer, S. & NicHerndon. Irys-Scaffolding: Irys-scaffolding Tools Version 1.0.0 https://doi.org/10.5281/zenodo.18591 (2015).
Zhang, J. et al. A high-density genetic map of tetraploid Salix matsudana using specific length amplified fragment sequencing (SLAF-seq). PLoS ONE 11, e0157777 (2016).
Hou, J. et al. Major chromosomal rearrangements distinguish willow and poplar after the ancestral “Salicoid” genome duplication. Genome Biol. Evol. 8, 1868–1875 (2016).
Hou, J., Wei, S., Pan, H., Zhuge, Q. & Yin, T. Uneven selection pressure accelerating divergence of Populus and Salix. Hortic. Res. 6, 37 (2019).
Qamar, M. T. et al. ppsPCP: a plant presence/absence variants scanner and pan-genome construction pipeline. Bioinformatics 35, 4156–4158 (2019).
Thibault, J. Nuclear DNA amount in pure species and hybrid willows (Salix): a flow cytometric investigation. Can. J. Bot. 76, 157–165 (1998).
Serapiglia, M. J., Gouker, F. E. & Smart, L. B. Early selection of novel triploid hybrids of shrub willow with improved biomass yield relative to diploids. BMC Plant Biol. 14, 74 (2014).
Horjales, M. Cantidades de DNA nuclear en árboles y arbustos. Nova Acta Cient. Compostel. (Bioloxía) 13, 23–33 (2003).
Zhang, M. L. A preliminary cladistic study on the multistaminal willows (Salix) in China. Bull. Bot. Res. 3, 299–305 (1994).
Zhang, J. et al. Determination of chromosome ploidy and genome size of Salix matsudana based on flow cytometry. Jiangsu Agr. Sci. 46, 40–43 (2018).
Ramsey, J. & Schemske, D. W. Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu. Rev. Ecol. Syst. 29, 467–501 (1998).
Doyle, J. J. & Egan, A. N. Dating the origins of polyploidy events. N. Phytol. 186, 73–85 (2010).
Haider, N. The origin of the B-genome of bread wheat (Triticum aestivum L.). Russ. J. Genet. 49, 263–274 (2013).
Sattler, M. C., Carvalho, C. R. & Clarindo, W. R. The polyploidy and its key role in plant breeding. Planta 243, 281–296 (2016).
Tu, Z. Y. Willow Breeding And Cultivation (Jiangsu Science & Technology Press, Nanjing, 1982).
Branco, R. & Masle, J. Systemic signalling through translationally controlled tumour protein controls lateral root formation in Arabidopsis. J. Exp. Bot. 70, 3927–3940 (2019).
Toscano-Morales, R., Xoconostle-Cázares, B., Martínez-Navarro, A. C. & Ruiz-Medrano, R. AtTCTP2 mRNA and protein movement correlates with formation of adventitious roots in tobacco. Plant Signal Behav. 11, e1071003 (2016).
Li, H. et al. A WD40 domain cyclophilin interacts with histone H3 and functions in gene repression and organogenesis in Arabidopsis. Plant Cell. 19, 2403–2416 (2007).
Yang, H. M. GENOMICS (China Science Press, 2016).
Boucher, L. D., Manchester, S. R. & Judd, W. S. An extinct genus of Salicaceae based on twigs with attached flowers, fruits, and foliage from the Eocene Green River Formation of Utah and Colorado, USA. Am. J. Bot. 90, 1389–1399 (2003).
Manchester, S. R., Judd, W. S. & Handley, B. Foliage and fruits of early poplars (Salicaceae: Populus) from the Eocene of Utah, Colorado, and Wyoming. Int. J. Plant Sci. 167, 897–908 (2006).
Wu, J. et al. Phylogeny of Salix subgenus Salix sl (Salicaceae): delimitation, biogeography, and reticulate evolution. BMC Evol. Biol. 15, 31 (2015).
Hohmann, N., Wolf, E. M., Lysak, M. A. & Koch, M. A. A time-calibrated road map of Brassicaceae species radiation and evolutionary history. Plant Cell. 27, 2770–2784 (2015).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199 (2010).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158 (2005).
Wicker, T. et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982 (2007).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Tarailo Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Prot. Bioinformatics. 25, 4–10 (2009).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, i215–i225 (2003).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr. Prot. Bioinformatics. 18, 3–4 (2007).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M. & Buell, C. R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics. 7, 327 (2006).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 (2010).
Tatusov, R. L. et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29, 22–28 (2001).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Dimmer, E. C. et al. The UniProt-GO annotation database in 2011. Nucleic Acids Res. 40, D565–D570 (2012).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Dan, S. P. SIGNAL SCAN: a computer program that scans DNA sequences for eukaryotic transcriptional elements. Comput. Appl. Bio. 7, 203 (1991).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31971681), Jiangsu Province Forestry Science and Technology Innovation and Promotion Project (LYKJ [2018]36) and Nantong University Scientific Research Start-up Project for Introducing Talents (18R08).
Author information
Authors and Affiliations
Contributions
J.Z. and J.C.X. conceived and designed the project. Y.J.L. and Y.N.J. performed the experiments. Y.H.C. and G.Y.L. conducted the gene expression analysis. M.X.Y., C.M.Y., B.L.L., and F.Z. analyzed the data. H.W.Y. wrote the paper. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Yuan, H., Li, Y. et al. Genome sequencing and phylogenetic analysis of allotetraploid Salix matsudana Koidz. Hortic Res 7, 201 (2020). https://doi.org/10.1038/s41438-020-00424-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-00424-8
This article is cited by
-
Comprehensive analysis of annexin gene family and its expression in response to branching architecture and salt stress in crape myrtle
BMC Plant Biology (2024)
-
Genomic and transcriptomic studies on flavonoid biosynthesis in Lagerstroemia indica
BMC Plant Biology (2024)
-
Overexpression of the Salix matsudana SmAP2-17 gene improves Arabidopsis salinity tolerance by enhancing the expression of SOS3 and ABI5
BMC Plant Biology (2022)
-
Conversion between duplicated genes generated by polyploidization contributes to the divergence of poplar and willow
BMC Plant Biology (2022)
