
Abstract
Snake gourd (Trichosanthes anguina L.), which belongs to the Cucurbitaceae family, is a popular ornamental and food crop species with medicinal value and is grown in many parts of the world. Although progress has been made in its genetic improvement, the organization, composition, and evolution of the snake gourd genome remain largely unknown. Here, we report a high-quality genome assembly for snake gourd, comprising 202 contigs, with a total size of 919.8 Mb and an N50 size of 20.1 Mb. These findings indicate that snake gourd has one of the largest genomes of Cucurbitaceae species sequenced to date. The snake gourd genome assembly harbors 22,874 protein-coding genes and 80.0% of the genome consists of repetitive sequences. Phylogenetic analysis reveals that snake gourd is closely related to sponge gourd but diverged from their common ancestor ~33–47 million years ago. The genome sequence reported here serves as a valuable resource for snake gourd genetic research and comparative genomic studies in Cucurbitaceae and other plant species. In addition, fruit transcriptome analysis reveals the candidate genes related to quality traits during snake gourd fruit development and provides a basis for future research on snake gourd fruit development and ripening at the transcript level.
Similar content being viewed by others


Introduction
As the second largest vegetable family, the Cucurbitaceae family comprises one of the most genetically diverse groups of plants. The members of this family are prevalent in tropical regions1,2 and many species are now cultivated worldwide as food crops. Snake gourd (Trichosanthes anguina L.; 2n = 2× = 22) is a diploid annual woody climber (liana) of the genus Trichosanthes in the family Cucurbitaceae2. This species is commonly called viper gourd, snake tomato, or long tomato, and has several synonyms (e.g., Trichosanthes cucumerina L.). Snake gourd originated in India or the Indo-Malayan region in tropical Asia3,4 and is widely distributed in Asian countries5,6,7. Its green, tender stems, leaves, and fruits are consumed as edible vegetables2,6, which have high nutritional value, because they are rich in vitamins, essential minerals, dietary fiber, and other nutrients, and are a wholesome, healthy addition to diets7,8. The fruits of snake gourd are frequently consumed when immature. As the fruits mature, the rind and flesh turn red, and the red flesh is used as a tomato substitute2. Snake gourd fruits can grow to 1.5 m in length, are serpentine in shape (hence the name snake gourd), and are used for ornamental purposes when they are mature. Their functional constituents include flavonoids, β-carotenoids, lycopene, and phenolic acids, which have beneficial pharmacological and therapeutic effects for humans2,9,10,11. For these reasons, snake gourd is a potentially economically important crop species with food, medicinal and ornamental value, and is worthy of further study and scientific research.
In the past decade, owing to the rapid advances in sequencing technology and bioinformatic algorithms, reference genomes of a number of cucurbit species have been assembled, including cucumber12,13,14,15,16,17, melon18,19,20, zucchini21, bottle gourd22, watermelon23,24,25, pumpkin26,27, wax gourd28, sponge gourd29, and bitter gourd30. However, no reference genome of snake gourd is available. Recent studies have shown that there is likely a common tetraploid ancestor of Cucurbitaceae species, and it has been inferred that cucurbits diverged from their common ancestor with grape 107–121 million years ago (Mya)31. Watermelon is believed to have differentiated from within the Cucurbitaceae family ~20.4 Mya and the divergence between melon and cucumber occurred 9.0–10.2 Mya26,32,33. However, the evolutionary history of the snake gourd genome remains largely unexplored.
In this study, we assembled the genome of snake gourd using Nanopore long reads combined with Hi-C chromatin interaction maps. Protein-coding genes and noncoding RNAs (ncRNAs) were predicted from the genome assembly, after which the predicted genes were functionally annotated. Phylogenetic and comparative genomic analyses indicated that snake gourd is closely related to sponge gourd. Transcriptome analysis revealed candidate genes that are involved in fruit texture, pigment accumulation, plant hormones, and resistance, and the expression profiles during fruit development and ripening, which may contribute to the unique characteristics of snake gourd fruits. The genome sequence presented in this study provides insights into both the structural characteristics of the snake gourd genome and the evolutionary relationship of snake gourd and related species and serves as a valuable resource for genomic research and comparative genomics within the Cucurbitaceae.
Results
Genome assembly, anchoring, and quality evaluation
For this study, snake gourd plants were grown in a greenhouse, as shown in Fig. 1a. The immature fruits were greenish-white, long and slender (the length could reach 1.5–2 m). Figure 1b shows a longitudinal section of a snake gourd and the flesh of the immature fruit is white and tough. Two Illumina libraries with fragment sizes of ~350 bp were constructed for snake gourd and sequenced on the Illumina Novaseq 6000 sequencing platform (Illumina, 9885 Towne Centre Drive, San Diego, CA 92121, USA) in paired-end mode and with a read length of 150 bp. After cleaning, 65.5 Gb of high-quality data were obtained, representing a 63.6-fold depth of the snake gourd genome, which has an estimated size of 1.03 Gb based on the K-mer depth distribution analysis of the paired-end Illumina reads. The snake gourd genome is relatively large, highly homozygous, and estimated to contain 70.5% repetitive sequences, with a heterozygosity level of ~0.02% and a GC content of ~7.1%.

a Snake gourd plants growing in the greenhouse of the Shouguang vegetable expo garden. b Longitudinal section of an immature snake gourd fruit
A nanopore library was constructed and sequenced, generating a total of 135.9 Gb of raw data. After cleaning, the data were reduced to 111.8 Gb, representing ~108.5× the total data composing snake gourd genome, with an N50 read length of 32.7 kb. De novo assembly of the Nanopore reads resulted in an initial assembly of 919.8 Mb consisting of 167 contigs with an N50 length of 21.9 Mb (Table 1). The single-base error rate in the genome sequence using Nanopore sequencing technology was 0.00156%. After error correction with Illumina paired-end and Hi-C interaction maps, we obtained a final assembly that was 919.8 Mb in size comprising 202 contigs and 69 scaffolds, with a contig N50 of 20.11 Mb and a scaffold N50 of 82.12 Mb (Table 1). A chromosome interaction heatmap was constructed (Fig. 2a), which showed a pattern consistent with that of the main Hi-C genome assemblies and provided confidence in the pseudomolecule construction. Based on the Hi-C contact maps, a total of 197 sequences covering ~918.8 Mb (99.89% of the assembled genome) clustered into 11 groups that correspond to the 11 chromosomes of snake gourd (Fig. 2b), with the longest being 118.8 Mb and the shortest being 64.0 Mb (Supplementary Table 1 and Table 1).

a Hi-C interaction heatmap of the snake gourd genome. The assembled genome was divided into 100 kb bins and valid interaction links of the Hi-C data were calculated between each pair of bins. The binary logarithm of the number of links in each bin was coded using colors ranging from light yellow to dark red, indicating the strength of Hi-C interaction links from low to high, respectively. LG01–LG11 represent the 11 chromosomes. b Landscape of the snake gourd genome. A: distribution of GC content (green); B: density of repeat sequences (blue); C: gene density (orange); D: noncoding RNA density (pink); E: syntenic blocks within the genome
To evaluate the quality of the assembly, we mapped the cleaned Illumina short reads to the assembled genome using BWA34, which had a mapping rate of 99.4%. Further evaluation using BUSCO (https://busco.ezlab.org/) with a database of 2,326 conserved core plant genes indicated that 95.7% of the core genes were found to be complete in the snake gourd genome assembly, including 93.5% that were single copies and 2.2% that were duplicated copies. In addition, 1.1% were found to be fragmented and only 3.2% were missing. Taken together, these results supported the integrity and high quality of the assembled snake gourd genome.
Repeat sequence annotation and gene prediction
Based on the principles of structure prediction and de novo prediction, we constructed a repeat sequence database of the snake gourd genome, which was used to screen the assembly for repeat sequences. A total of 736.1 Mb (80.0%) of repetitive sequences were identified in the assembled genome. Among these repetitive sequences, long terminal repeat (LTR) retrotransposons were predominant, constituting 66.7% of the snake gourd genome assembly, with 45.6% belonging to the copia type and 19.8% belonging to the gypsy type (Supplementary Table 2).
Three prediction methods—an ab initio strategy, a homology-based strategy, and an RNA sequencing (RNA-seq) strategy—were used to predict protein-coding genes in the snake gourd genome and the predictions from these three methods were integrated using EVM35 v1.1.1 software (Supplementary Fig. 1). Ultimately, 22,874 protein-coding genes with a total length of 121.4 Mb were predicted in the genome (Table 2) and were functionally annotated, the annotations of which are shown in Supplementary Table 3. Of the predicted genes, a total of 22,129 (96.74%) were annotated by five functional databases (Table 2). In addition, we also identified 74 miRNAs, 294 rRNAs, 1,167 tRNAs, and 3,021 pseudogenes.
Evolution of the snake gourd genome
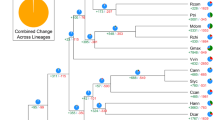
To understand the evolution of the snake gourd genome, we collected genome sequences of representative plant species and performed a comparative genomic analysis with the genome sequence of snake gourd. Genes from 13 selected plant species, including 9 cucurbits (T. anguina L., Luffa cylindrica L., Cucumis sativus L., Cucumis melo, Citrullus lanatus, Lagenaria siceraria, Cucurbita moschata, Cucurbita pepo, and Momordica charantia), two rosid species (Vitis vinifera L. and Arabidopsis thaliana), one monocot (Oryza sativa), and one in the basal lineage of angiosperms (Amborella trichopoda), clustered into 34,827 gene families. A total of 17,057 gene families were identified in snake gourd, 125 of which (comprising 451 genes) were specific to the snake gourd genome (Supplementary Table 4). Clustering analysis revealed a total of 14,148 single-copy genes in snake gourd, accounting for 61.9% of the predicted genes, which is similar to that in other cucurbit species, such as L. cylindrica, C. sativus, C. melo, C. lanatus, L. siceraria, and M. charantia, in which no recent whole-genome duplication (WGD) events occurred, but is substantially higher than that in C. moschata and C. pepo, whose genomes underwent a recent WGD event (Fig. 3a). The clustering of gene families in snake gourd and several other cucurbit species, including sponge gourd, watermelon, bottle gourd, and pumpkin, is shown in Fig. 3b and indicates that the number of snake gourd gene families (17,057) was most similar to the number of sponge gourd gene families (16,986).

a Gene copy number distribution in snake gourd and 12 other plant species. b Venn diagram representing the clusters of gene families in snake gourd and four other related cucurbit species (sponge gourd, watermelon, bottle gourd, and pumpkin). c Phylogenetic tree of snake gourd and 12 other species based on a concatenated alignment of 970 single-copy protein sequences. The tree is rooted with A. trichopoda as the outgroup. CAFÉ-based estimates of gene family expansions and contractions. The red and blue numbers indicate expanded and contracted gene families, respectively. d Ks distribution in snake gourd and other representative plant species
In addition, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed for gene families specific to snake gourd. GO analysis showed that these families were enriched in genes involved in metabolic processes, cellular processes, and biological regulation or encoded products that have binding, nucleic acid binding, DNA binding, and catalytic activity function (Supplementary Fig. 2b). KEGG pathway analysis indicated that genes related to flavonoid biosynthesis, regulation of autophagy, fatty acid degradation, etc., were enriched in the snake gourd-specific families (Supplementary Fig. 2a). These included two genes (EVM0012390.1 and EVM0020640.1) encoding flavonol synthase/flavanone 3-hydroxylase in the flavonoid biosynthetic pathway, two autophagy-related genes associated with the regulation of the autophagy pathway, and two genes encoding alcohol dehydrogenases (ADHs) (EVM0001187.1 and EVM0017122.1), which are involved in fatty acid degradation, tyrosine metabolism, α-linolenic acid metabolism, and glycolysis/gluconeogenesis. Furthermore, a number of other genes were identified that are known to participate in fruit ripening, such as those encoding cell wall-associated hydrolase, phenylalanine ammonia-lyase (PAL) 1, malonate–CoA ligase, and aquaporin PIP2-2. Together, these specific genes and gene families may contribute to the unique fruit features of snake gourd (Table 3).
Based on 970 single-copy genes in snake gourd and in 12 other plant species, a phylogenetic tree was constructed. The phylogenetic tree showed that, of all the Cucurbitaceae species examined, snake gourd is most closely related to sponge gourd (Fig. 3c). Snake gourd and sponge gourd diverged from their common ancestor approximately 33–47 Mya after the divergence from the common ancestor together with bitter gourd (44–87 Mya) but before the divergence between watermelon and bottle gourd (14–27 Mya) (Fig. 3c). In addition, we performed a comparative analysis of the evolution of gene families in the 13 plant species. In snake gourd, 226 gene families comprising 1406 genes exhibited significant expansion (p < 0.01) relative to those of the last common ancestor, whereas 268 gene families showed contraction (Fig. 3c). KEGG and GO functional analyses showed that most of the members of the expanded snake gourd gene families are involved in metabolic processes, cellular processes, cell catalytic activity, binding, flavonoid biosynthesis, regulation of autophagy, fatty acid degradation, and tyrosine metabolism (Supplementary Fig. 3), and included genes encoding cellulose synthase, wall-associated receptor kinase, exopolygalacturonase, polygalacturonase (PG), lipoxygenase, linoleate 9S-lipoxygenase 6, terpene synthase, (−)-germacrene d-synthase, (+)-γ-cadinene synthase, myrcene synthase, 1-aminocyclopropane-1-carboxylate oxidase (ACO), auxin-induced protein 22D, 23 kDa jasmonate-induced protein, anthocyanidin 3-O-glucosyltransferase, and flavonoid hydroxylase. These genes are involved in cell wall biosynthesis and degradation, flavor and aromatic compound generation, phytohormone synthesis and signal transduction, and flavonoid biosynthesis. Other gene families involved in plant defense and response to pathogens were also found to be expanded in snake gourd, such as those whose members encode UDP-glucosyltransferase, salicylate carboxymethyltransferase, tobacco mosaic virus (TMV) resistance protein N, and other disease resistance proteins.
WGD events have occurred widely in angiosperms and many plant species have experienced genome duplications in their evolutionary history, which are of great significance in understanding speciation, genome evolution, and gene neofunctionalization36. However, our in-depth genomic analysis indicated that no recent WGD events have occurred in the snake gourd genome, which is consistent with the findings of a recent report37. The Ks distribution among these species suggested that snake gourd diverged from sponge gourd ~42 Mya and from watermelon ~47 Mya (Fig. 3d). A peak centering on a Ks of ~1.32 was observed between snake gourd paralogous pairs (Fig. 3d), which corresponds to the ancient whole-genome triplication (the γ event) shared by all core eudicots31. In addition, genome collinearity analysis between snake gourd, sponge gourd, and watermelon (Supplementary Fig. 4) showed a high degree of gene-order conservation with the snake gourd genome.
Transcriptomes at different ripening stages of snake gourd fruits
To identify potential candidate genes related to fruit characteristics and quality during fruit development and ripening, transcriptome analysis was used to study changes in differentially expressed genes (DEGs) in the fruits at different stages. The results showed that there were 362 common DEGs in the development and ripening stages (20 d vs. 40 d and 40 d vs. 60 d) (Fig. 4a). Among them, five genes related to disease resistance and defense responses overlapped between the two groups, leucine-rich repeat receptor-like protein kinase TDR, LRR receptor-like serine/threonine-protein kinase FEI 1, TMV resistance protein N, leucine-rich repeat extensin-like protein 6, and respiratory burst oxidase homolog protein E, all of which participate in the plant–pathogen interaction pathway (Supplementary Table 5). These genes were upregulated at 20 d vs. 40 d and downregulated at 40 d vs. 60 d, which may be related to changes in fruit resistance during ripening.

a Venn diagram representing the number of DEGs between the 20 d vs. 40 d and 40 d vs. 60 d groups; 362 DEGs overlapped between the two groups. b Upregulated KEGG pathways in the 20 d vs. 40 d group comparison. c Upregulated GO terms related to biological processes in the 20 d vs. 40 d group comparison. d Netplot of significantly enriched GO terms in the 20 d vs. 40 d group comparison. e Upregulated KEGG pathways in the 40 d vs. 60 d group comparison
During the period of fruit development, a total of 480 genes, including 356 (74%) upregulated and 124 (26%) downregulated genes, were differentially expressed in the 20 d vs. 40 d comparison. KEGG pathway analysis of the upregulated genes showed that they were mainly enriched in the pathways involved in phenylpropanoid biosynthesis, amino sugar and nucleotide sugar metabolism, protein processing in the endoplasmic reticulum and starch and sucrose metabolism (Fig. 4b). The expression of several peroxidase genes, such as peroxidase 31 (increase of 3.4-fold), peroxidase 55-like (increase of 4.0-fold), peroxidase 5 (increase of 6.4-fold), peroxidase 64-like (increase of 8.8-fold), peroxidase 2-like (increase of 11.6-fold), peroxidase 21-like (increase of 10.5-fold), and probable mannitol dehydrogenase (increase of 2.1-fold), markedly changed in snake gourd fruit (Supplementary Table 5). Of the upregulated GO terms, the most enriched categories related to biological processes included the xyloglucan metabolic process, cell wall biogenesis, the carbohydrate metabolic process, and cell wall organization (Fig. 4c). The corresponding gene networks associated with significantly abundant GO items in the 20 d vs. 40 d group are shown in Fig. 4d. Many enzymes related to cell wall metabolism were identified as being upregulated, including β-glucosidase (BGLU) 18-like, β-galactosidase (GAL) 5-like/10, pectinesterase-like (PE), expansin-A4/A10-like (EXP), endoglucanase (EG) 10/11/17-like, glucan endo-1,3-β-d-glucosidase, and glucan endo-1,3-β-glucosidase 13-like but not cellulose synthase-like protein H1. In addition, increased expression of plant hormone-related genes was also found, including abscisic acid receptor PYR1/PYL4-like, auxin-induced protein 22D-like, and auxin-responsive protein IAA16/IAA21-like/IAA29-like. It is generally known that the phytohormone auxin can regulate cell growth and induce tracheary element differentiation38.
We identified 4801 genes that were differentially expressed by comparing the transcriptome at 60 d with that at 40 d. A total of 1957 (41%) genes were upregulated and 2844 (59%) genes were downregulated. In specific gene families of snake gourd, five genes were found to be differentially expressed: PAL, cysteine protein inhibitor 1-like, MLP-like protein 329, poly transporter 5-like, and PAL 3. Their expression profiles were found to be downregulated after 60 d compared with 40 d. The results of the KEGG pathway analysis of the upregulated genes suggested that the most enriched pathways were involved in fatty acid biosynthesis, carbon metabolism, pyruvate metabolism, and carotenoid biosynthesis (Fig. 4e). Among the 15 genes involved in carotenoid synthesis, 10 were upregulated, including phytoene synthase (PSY) (increase of 1.7-fold), 15-cis-phytoene desaturase (increase of 4.6-fold), 15-cis-zeta-carotene isomerase (increase of 5.1-fold), zeta-carotene desaturase (ZDS) (increase of 19.0-fold), prolycopene isomerase (CRTISO) (increase of 25-fold), and β-carotene 3-hydroxylase 1 (increase of 40.0-fold), and 5 were downregulated, including lycopene epsilon cyclase, lycopene β-cyclase, probable 9-cis-epoxycarotenoid dioxygenase NCED5, and abscisic acid 8-hydroxylase 4-like. Moreover, 11 genes related to the accumulation of flavonoids and anthocyanidins in the fruits were also found, including anthocyanidin 3-O-glucosyltransferase 2-like, flavonoid 3-monooxygenase-like, flavonoid 3,5-methyltransferase-like, flavonol synthase/flavanone 3-hydroxylase-like, and isoflavone reductase-like protein, which exhibited biphasic expression patterns. PAL is involved in the phenylalanine metabolism pathway and functions prior to lignin synthesis, and the expression of many peroxidases related to enzymes associated with lignification, the cell wall, auxin catabolism, defense against pathogens, and scavenging of reactive oxygen species is downregulated. In addition, the expression levels of numerous genes related to plant hormones synthesis and signal transduction were significantly altered: the levels of ACO homolog 1/3-like, ethylene receptor 2-like, ethylene response sensor 1, the serine/threonine-protein kinase CTR1, ethylene insensitive 3-like 3 protein, auxin response factor 18 (ARF), auxin-responsive protein SAUR71-like, gibberellin-regulated protein 1-like (GRP), and indole-3-acetic acid-amido synthetase GH3.17 were upregulated, and the expression levels of others, such as ACO1/3, ethylene-responsive transcription factor 1B-like (ERF), indole-3-acetic acid-amido synthetase GH3.10/GH3.6-like, auxin-induced protein 15A/22D/22B-like, ARF 4/9/18, auxin-responsive protein IAA/SAUR50-like, auxin transporter-like protein 1/2, the jasmonic acid-amido synthetase JAR1, abscisic acid receptor PYR1-like, GRP 9/4-like, and the AP2-like ethylene-responsive transcription factor ANT, were downregulated. Furthermore, the mRNAs for some enzymes related to fruit cell wall structure also changed significantly, with PE, PE 3-like, pectate lyase 5/8/12/18 (PL), pectin acetylesterase 8-like, PG-like, PG At1g48100-like, PG QRT3-like, GAL 3/10/5-like, BGLU 18/44-like, glucan endo-1,3-β-glucosidase, expansin (EXP), EG 6/10/11/17/24-like, and cellulose synthase-like protein G3 downregulated while others such as β-d-xylosidase1-like, GAL 13-like, GAL 16, GAL-like, cellulose synthase-like protein E1/H1, and EG 9-like were downregulated. These changes in mRNAs of cell wall enzymes may be related to fruit texture changes, as the fruits were softer after 60 d compared with 40 d.
We focused our subsequent analysis on cell wall-related gene families whose members are responsible for fruit texture in snake gourd. The glycosyl hydrolase family (GH) related to cell wall structure was selected to construct a phylogenetic tree. The GH families were divided into four subfamilies: GH-1, GH-9, GH-28, and GH-35. The members of the GH-28 and GH-35 subfamilies encode the majority of GH proteins. The genes belonging to the GH-28 and GH-35 subfamilies in snake gourd showed the closest phylogenetic relationships with those of pumpkin and sponge gourd (Fig. 5a). In the snake gourd genome, the GH-1 subfamily contained five members, but only one, one, and three proteins belonged to the GH-1 subfamily in Arabidopsis, pumpkin, and sponge gourd, respectively; moreover, there were no proteins that clustered within the GH-9 subfamily in these three plant species (Fig. 5a). Based on the transcriptome data, we further analyzed the expression of 15 DEGs related to cell wall modification and pigment accumulation in the 20 d vs. 40 d (SG1 vs. SG2) and 40 d vs. 60 d (SG2 vs. SG3) group comparisons, respectively. A heatmap of these DEGs showed the gene expression profiles at different stages (Fig. 5b, c). In the 20 d vs. 40 d group comparison (SG1 vs. SG2), numerous members of the GH family were highly expressed at this fruit development stage, with the exception of PE (EVM0004218), and the results of the three replications were similar (Fig. 5c). In the 40 d vs. 60 d group comparison (SG2 vs. SG3), ZDS (EVM0002371), prolycopene isomerase (EVM0010946), and 9-cis-epoxycarotenoid dioxygenase NCED2 (EVM0012548) had much higher expression in SG3, whereas the expression levels of abscisic acid 8-hydroxylase 4-like (EVM0017015 and EVM0004622) and 9-cis-epoxycarotenoid dioxygenase NCED5 (EVM0015273) decreased during fruit maturation (Fig. 5c).

a Phylogenetic tree of glycosyl hydrolase families among snake gourd, sponge gourd, Arabidopsis, and pumpkin. b Heatmap showing the expression of fruit texture-related genes at the 20 d (SG1) and 40 d (SG2) stages. c Heatmap showing the expression of fruit pigment accumulation-related genes at the 40 d (SG2) and 60 d (SG3) stages. The red and green colors indicate high and low expression levels, respectively
Discussion
Snake gourd is an excellent vegetable species that is popular for both its unique flavor and its nutritional, health, medicinal and ornamental value and has high utilization potential2. However, research on this species is limited3, and related molecular-level studies are scarce. We aimed to sequence, assemble, annotate, and comparatively analyze the snake gourd genome to provide a basis for future investigations and improvement of this species. This is the first report of a high-quality genome for snake gourd, which has one of the largest genomes of Cucurbitaceae species sequenced to date.
The assembled genome size is ~919.76 Mb, which is close to that of wax gourd (913 Mb) but larger than those of other Cucurbitaceae species, such as watermelon, melon, pumpkin, bottle gourd, cucumber, zucchini, bitter gourd (200–400 Mb), and sponge gourd (669 Mb)17,18,19,20,21,22,23,24,25,26,27,28,29,30 (Table 4). We predicted 22,874 protein-coding genes, which is similar to the number of genes present in melon19, slightly more than the number in bottle gourd22, but less than the number in the eleven other Cucurbitaceae species examined (Table 4). Repetitive sequences such as transposable elements play crucial roles in genome evolution, chromosome rearrangement, and gene regulation39 but have presented a major challenge for producing high-quality genome assemblies. We found that 80.0% of the snake gourd genome is composed of repeated sequences, with LTRs accounting for 66.7% of the genome, which is quite high compared with the percentage of repeat content in other Cucurbitaceae genomes. BUSCO (https://busco.ezlab.org/)-based assessment revealed that the snake gourd assembly is more complete than or comparable to other reported cucurbit reference genomes (Table 4) and provides valuable information about evolutionary events and gene family expansion in snake gourd; in addition, this assembly serves as a useful resource for comparative genomic studies in Cucurbitaceae.
In specific gene families of snake gourd, a number of genes have been found to potentially participate in fruit ripening, such as several genes encoding autophagy-related proteins, ADHs, and the aquaporin PIP2-2. Autophagy is involved in plant processes such as floret ripening (in wheat)40, root tip cell growth and differentiation41, and chloroplast degradation in senescing leaves42. The autophagy pathway has also been reported to be involved in plant innate immunity43 and responses to a variety of abiotic stresses44, and in assisting plants in surviving nutrient-limitation stress45. ADH enzymes play multiple roles in anaerobic fermentation, aerobic fermentation, and the production of scents that discourage predation, attract pollinators, and facilitate seed dispersal46. In petunia, both the ADH2 and ADH3 genes act in the lipoxygenase pathway to produce floral scents47 and flavor volatiles in ripening fruits. A previous study reported a strawberry fruit-specific aquaporin, FaPIP1;1, which showed an expression profile associated with fruit ripening48. Further study indicated that the FaPIP aquaporins showed an expression pattern associated with fruit firmness48. In addition, several specific genes were found to be differentially expressed between 40 d and 60 d, which may contribute to the unique snake gourd fruit features.
Between 20 d and 40 d, the snake gourd fruits became longer and harder, and we detected increased transcript levels of genes related to cell division, cell expansion, fruit growth, and protection of the fruit surface49. We identified transcripts for highly expressed cell wall-modifying genes similar to those expressed in expanding cucumber50,51, tomato52, melon53, and watermelon54 fruits, such as EXPs, endo-1,2-B-glucanase, BGLUs, PLs, and pectin methylesterases. Major increases in transcripts for mRNAs encoding enzymes related to cell wall metabolism were detected, including GAL 10 (increase of 14.3-fold), PE-like (increase of 21.6-fold), EG 17-like (increase of 21.2-fold), expansin-A10-like (increase of 27.1-fold), UDP-glucuronate 4-epimerase 1-like (increase of 29.8-fold), and PE 53 (increase of 30.4-fold), which may be involved in the production of longer and harder fruits. We also detected the upregulated expression of auxin-related genes. These genes may be involved in fruit elongation, as many studies have shown that phytohormones, especially auxin and gibberellins, are related to fruit growth and changes in fruit shape55. These results are consistent with those of previous studies that found that three homologs of auxin-related genes were differentially expressed in developing watermelon fruits54. In addition, we found an increase in resistance (R) gene transcripts, such as those of TMV resistance protein N, leucine-rich repeat extensin-like protein 3, and LRR receptor-like serine/threonine-protein kinase FEI 1. Studies have shown that younger and smaller fruits are more susceptible to infection than older and larger fruits56, and changes in the expression of resistance genes may explain the stronger resistance of larger fruits compared with smaller ones. These genes mainly regulate the resistance of plants to pathogens and insects, and in cucumber, eight DEGs have been identified that may be associated with aphid resistance57.
Transcripts for several enzymes mentioned above related to cell wall metabolism were found to be downregulated during ripening from 40 to 60 d, which may be the cause of fruit softening at 60 d. The expression levels of these enzymes are different among various fruits and at different fruit developmental stages. Studies on watermelon have shown that there are differences in the expression of genes related to cell wall components in two kinds of flesh with different textures58, and the key genes involved in the regulation of flesh texture were differentially expressed in cultivated and wild watermelon59. In addition, the genes involved in the upstream and downstream steps of the carotenoid biosynthesis or degradation pathway were upregulated and downregulated, respectively. These changes may contribute to the increased carotenoid accumulation, making the fruit turn orange after ripening. This is supported by research on carotenoid accumulation and related gene expression during squash fruit development60.
Materials and methods
Snake gourd sample collection and genome sequencing
Young fresh leaf samples were collected from snake gourd plants grown in the greenhouse of the Shouguang vegetable expo garden. Genomic DNA was extracted from young leaves and used to construct two Illumina DNA libraries whose fragment size was ~350 bp, according to the standard protocols provided by the Illumina company. The libraries were sequenced on an Illumina HiSeq X platform in paired-end mode and with read length of 150 bp. The sequence data were evaluated (GC distribution statistics and quality value assessment) and filtered to obtain high-quality clean reads, which were subsequently used for estimation of the genome size, GC content, heterozygosity level, and postassembly error correction and evaluation. The raw sequencing reads were processed for quality control and for the removal of adapter and low-quality sequences using a custom Perl script. After trimming, reads with lengths less than 100 bp or a Q30 value < 85% were discarded.
A Nanopore library was constructed from high-molecular-weight DNA using a Ligation Sequencing Kit (SQK-LSK109) and sequenced on a PromethION R9 flow cell with a PromethION Flow Cell Priming Kit (EXP-FLP001.PRO.6) (Oxford Nanopore Technologies, UK) according to the manufacturer’s instructions. The Oxford Nanopore reads were self-corrected using Canu61 and the corrected reads were assembled into contigs using SMARTdenovo (https://github.com/ruanjue/smartdenovo). The assembled contigs were further polished using the Racon62 and Pilon63 programs in conjunction with cleaned Illumina reads.
We also constructed Hi-C fragment libraries (insert size of 300–700 bp) and sequenced them using the Illumina platform. The raw Hi-C reads were processed to trim adapter sequences and to remove low-quality reads. The cleaned Hi-C reads were first truncated at the putative Hi-C junctions, and then the resulting trimmed reads were aligned to the genome assembly with BWA34 (version 0.7.10-r789). Only uniquely aligned paired-end reads with a mapping quality greater than 20 were used for further analysis. Invalid read pairs including dangling-end and self-cycle, religation and dumped products were filtered by HiC-Pro64 v2.8.1. Approximately 75% of the uniquely mapped read pairs were valid interaction pairs and were used for correction of the assembled contigs, which were then clustered, ordered, and oriented onto chromosomes by LACHESIS65 with the parameters “CLUSTER_MIN_RE_SITES, 52; CLUSTER_MAX_LINK_DENSITY, 2; ORDER_MIN_N_RES_IN_TRUN, 51; ORDER_MIN_N_RES_IN_SHREDS, 50.”
Gene prediction and functional annotation
A repetitive sequence database of the snake gourd genome was constructed using LTR_FINDER66 and RepeatScout67. Repeat sequences in the database were classified using PASTEClassifier68 and then merged together with the repeats in RepBase69 to construct the final repetitive sequence database. Repetitive sequences of the snake gourd genome were predicted using RepeatMasker70 based on the constructed repetitive sequence database.
Protein-coding genes were predicted from the snake gourd genome using three different strategies: ab initio prediction, homology-based prediction, and transcript-based prediction. We used GENSCAN71, Augustus72 v2.4, GlimmerHMM73 v3.0.4, GeneID74 v1.4, and SNAP75 (version 2006-07-28) for ab initio predictions, and GeMoMa76 v1.3.1 was used for homology-based predictions using protein sequences from A. thaliana, C. lanatus, C. sativus, and L. cylindrica. For transcript-based predictions, a mixture of four tissues (root, stem, leaf and fruit tissues) of snake gourd was used to construct an Illumina RNA-Seq library, which was subsequently subjected to PE (2 × 150 bp) sequencing on an Illumina HiSeq X Ten platform (Illumina, San Diego, CA, USA). After discarding the reads with low-quality bases, adapter sequences, and duplicated sequences, the retained high-quality clean reads were mapped to the snake gourd genome assembly using HISAT277 v2.0.4 and, based on the mapping results, the reads were assembled into transcripts using StringTie78 v1.2.3, TransDecoder v2.0 (http://transdecoder.github.io) and GeneMarkS-T79 v5.1; the assembled transcripts were subsequently used for gene prediction. RNA-Seq reads were also de novo-assembled using Trinity80 and PASA81 v2.0.2 was used for gene prediction from these de novo-assembled transcripts. Finally, EVM35 v1.1.1 was used to integrate the prediction results obtained by the above three methods. In addition, we also predicted different ncRNAs. MicroRNA and rRNA sequences were predicted based on the comparison of the snake gourd genome to the content of the Rfam82 database via BLASTn and tRNA sequences were predicted using tRNAscan-SE83. Pseudogene prediction was also performed. Using the predicted protein sequence, through a BLAST84 comparison, we identified possible homologous gene sequences in the genome and then used GeneWise85 to identify premature termination codons and frameshift mutations in the gene sequences to identify pseudogenes.
The protein sequences of the snake gourd genes were compared to the content of the NCBI nr86, KOG87, GO88, KEGG89, and TrEMBL90 databases using BLAST91 v2.2.31, with an E-value cutoff of 1 × 10−5, and functional annotations of the snake gourd genes were derived from the homologous sequences in these databases.
Comparative genomic analysis
The protein sequences from snake gourd and 12 other representative plant species were clustered into orthologous groups using OrthoFinder92 v2.3.7. The obtained orthologous groups (gene families) were annotated using the PANTHER v15 database93. GO and KEGG enrichment analyses of gene families unique to snake gourd were ultimately performed using ClusterProfile94 v3.14.0.
By the use of 970 single-copy protein sequences, an evolutionary tree was constructed using the maximum likelihood method implemented in IQ-TREE95 v1.6.11, with A. trichopoda as the root and the number of bootstraps set to 1000. We then used MCMCTee software in the PAML package96 v4.9i to calculate the divergence time. The number of iterations of the Markov chain included a burn-in number of 700,000, a sampfreq of 30, and an nSample of 6,000,000.
Using the results of the evolutionary tree with differentiation time and gene family clustering by CAFE97 v4.2, we estimated the number of gene family members of each branch’s ancestor via a birth mortality model, which was used to predict the contraction and expansion of gene families of the snake gourd relative to its ancestors. We determined whether the expansion or contraction was significant using a P-value cutoff of 0.05. The expanded and contracted gene families identified in snake gourd were annotated with PANTHER, and GO and KEGG enrichment analyses on these families were performed with ClusterProfile.
Collinearity and WGD analyses
Diamond98 v0.9.29.130 was used to compare the protein sequences of two species and determine similar gene pairs (E-value < 1 × 10−5, C score > 0.5). Based on the diamond results, collinear blocks between the genomes of the two species were identified using MCScanX99.
WGD events were determined based on the distribution of the synonymous substitution rate (Ks) and fourfold degenerate (4DTv) sites of paralogous genes, which were calculated using WGD software100 in conjunction with a Perl script (https://github.com/JinfengChen/Scripts), respectively.
Transcriptome
Fresh snake gourd fruit samples at 20, 40, and 60 d were collected for transcriptome analysis. cDNA libraries were obtained by PCR enrichment. After the library was checked for quality by quantitative PCR, the Illumina platform was used for sequencing. The data were cleaned by removing low-quality sequences and those containing adapter reads and used for sequence alignment with the specified reference genome. The transcriptome was assembled using StringTie78. Differential expression analysis was performed between the different sample groups. Three replicates were used for each sample and DESeq2101 was used for differential expression analysis between sample groups to obtain the DEG sets between two biological samples. During the detection of DEGs, a fold change > 2 and a false discovery rate (FDR) < 0.01 were used. As a screening standard, the fold change (0.01) represents the FDR between two samples by the use of a corrected P-value denoting significant differences. The gene functions were annotated on the basis of the content from six databases: the Nr, Pfam, KOG/COG, Swiss-Prot, KEGG, and GO databases.
Phylogenetic tree construction
Predicted proteins from the genome were scanned using HMMER v3 (http://hmmer.org/download.html) employing the hidden Markov model (HMM) corresponding to the Pfam GH family. From the proteins obtained using the raw HMM, a high-quality protein set (E-value < 1 × 10 − 20 and manual verification of an intact domain) was aligned and used to construct a specific GH family HMM using hmmbuild from the HMMER v3 suite. With this new specific HMM, all proteins with an E-value lower than 0.01 were selected. The GH genes were further filtered based on manual curation and functional annotations (supplied by Beijing Biomarker).
Data availability
The raw genome and transcriptome sequencing data have been deposited into the NCBI Sequence Read Archive (SRA) database under BioProject accession numbers PRJNA640193 and PRJNA649380.
References
Schaefer, H. & Renner, S. S. Phylogenetic relationships in the order Cucurbitales and a new classification of the gourd family (Cucurbitaceae). Taxon 60, 122–138 (2011).
Devi, N. Medicinal values of Trichosanthus cucumerina L. (snake gourd) - a review. Br. J. Pharm. Res. 16, 1–10 (2017).
Adebooye, O. C. Phytoconstituents and anti-oxidant activity of the pulp of snake tomato (Tricosanthes cucumerina). Afr. J. Tradit. Complement. Altern. Med. 5, 173–179 (2008).
Roy, R. P., Saran, S. & Dutt, B. Cytogenetics of the cucurbitaceae. Dev. Plant Genet. Breed. 2, 181–199 (1991).
Arawwawala, M., Thabrew, I. & Arambewela, L. Antidiabetic activity of Trichosanthes cucumerina in normal and streptozotocin–induced diabetic rats. Int. J. Biol. Sci. 3, 287–296 (2009).
Liyanage, R., Nadeeshani, H., Jayathilake, C., Visvanathan, R. & Wimalasiri, S. Comparative analysis of nutritional and bioactive properties of aerial parts of snake gourd (Trichosanthes cucumerina Linn.). Int. J. Food Sci. 2016, 1–7 (2016).
Ojiako, O. A. & Igwe, C. U. The nutritive, anti-nutritive and hepatotoxic properties of Trichosanthes anguina (snake tomato) fruits from Nigeria. Pak. J. Nutr. 7, 85–89 (2008).
Rana, N. P. & Pandit, M. K. Studies on genetic variability, character association and path analysis in snake gourd (Trichosanthes anguina L.) genotypes. J. Crop Weed 7, 91–96 (2011).
Sandhya, S., Vinod, K. R., Sekhar, J. C., Aradhana, R. & Nath, V. S. An updated review on Tricosanthes cucumerina L. Int. J. Pharm. Sci. Rev. Res. 1, 56–60 (2010).
Yusuf, A. A., Folarin, O. M. & Bamiro, F. O. Chemical composition and functional properties of snake gourd (Tricosanthes cucumerina) seed flour. Niger. Food J. 25, 36–45 (2007).
Sathesh, K. S., Ravi, K. B. & Krishna, M. G. Hepatoprotective effect of Trichosanthes cucumerina Var cucumerina L. on carbon tetrachloride induced liver damage in rats. J. Ethnopharmacol. 123, 347–350 (2009).
Huang, S. et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41, 1275–1281 (2009).
Li, Z. et al. RNA-Seq improves annotation of protein-coding genes in the cucumber genome. BMC Genomics 12, 540 (2011).
Woycicki, R. et al. The genome sequence of the North-European cucumber (Cucumis sativus L.) unravels evolutionary adaptation mechanisms in plants. PLoS ONE 6, e22728 (2011).
Yang, L. et al. Chromosome rearrangements during domestication of cucumber as revealed by high-density genetic mapping and draft genome assembly. Plant J. 71, 895–906 (2012).
Qi, J. et al. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 45, 1510–1515 (2013).
Li, Q. et al. A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). GigaScience 8, giz072 (2019).
Garciamas, J. et al. The genome of melon (Cucumis melo L.). Proc. Natl Acad. Sci. USA 109, 11872–11877 (2012).
Zhang, H. et al. A high-quality melon genome assembly provides insights into genetic basis of fruit trait improvement. iScience 22, 16–27 (2019).
Castanera, R., Ruggieri, V., Pujol, M., Garcia-Mas, J. & Casacuberta, J. M. An improved melon reference genome with single-molecule sequencing uncovers a recent burst of transposable elements with potential impact on genes. Front. Plant Sci. 10, 1815 (2020).
Montero-Pau, J. et al. De novo assembly of the zucchini genome reveals a whole-genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnol. J. 16, 1161–1171 (2018).
Wu, S. et al. The bottle gourd genome provides insights into Cucurbitaceae evolution and facilitates mapping of a Papaya ring-spot virus resistance locus. Plant J. 92, 963–975 (2017).
Guo, S. et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45, 51–58 (2013).
Guo, S. et al. Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat. Genet. 51, 1616–1623 (2019).
Wu, S. et al. Genome of ‘Charleston Gray’, the principal American watermelon cultivar, and genetic characterization of 1,365 accessions in the U.S. National Plant Germplasm System watermelon collection. Plant Biotechnol. J. 17, 2246–2258 (2019).
Sun, H. et al. Karyotype stability and unbiased fractionation in the paleo-allotetraploid Cucurbita genomes. Mol. Plant 10, 1293–1306 (2017).
Barrera-Redondo, J. et al. The genome of Cucurbita argyrosperma (Silver-Seed Gourd) reveals faster rates of protein-coding gene and long noncoding RNA turnover and neofunctionalization within Cucurbita. Mol. Plant 12, 506–520 (2019).
Xie, D. et al. The wax gourd genomes offer insights into the genetic diversity and ancestral cucurbit karyotype. Nat. Commun. 10, 5158 (2019).
Zhang, T. et al. Long-read sequencing and de novo assembly of the sponge gourd cylindrica (L.) Roem. Genome Mol. Ecol. Resour. 20, 511–519 (2020).
Urasaki, N. et al. Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24, 51–58 (2016).
Wang, J. et al. An overlooked paleotetraploidization in cucurbitaceae. Mol. Biol. Evol. 35, 16–26 (2018).
Bellot, S. & Renner, S. S. Exploring new dating approaches for parasites: the worldwide Apodanthaceae (Cucurbitales) as an example. Mol. Phylogenet. Evol. 80, 1–10 (2014).
Schaefer, H., Heibl, C. & Renner, S. S. Gourds afloat: a dated phylogeny reveals an Asian origin of the gourd family (Cucurbitaceae) and numerous oversea dispersal events. Proc. R. Soc. B 276, 843–851 (2009).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, 1–22 (2008).
Van de Peer, Y., Maere, S. & Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 10, 725–732 (2009).
Guo, J. et al. Phylotranscriptomics in Cucurbitaceae reveal multiple whole genome duplications and key morphological and molecular innovations. Mol. Plant 13, 1117–1133 (2020).
Fukuda, H. Tracheary element differentiation. Plant Cell 9, 1147–1156 (1997).
Heitz, T., Bergey, D. R. & Ryan, C. A. A gene encoding a chloroplast-targeted lipoxygenase in tomato leaves is transiently induced by-wounding, systemin, and methyl jasmonate. Plant Physiol. 114, 1085–1093 (1997).
Ghiglione, H. O. et al. Autophagy regulated by day length determines the number of fertile florets in wheat. Plant J. 55, 1010–1024 (2008).
Yano, K., Suzuki, T. & Moriyasu, Y. Constitutive autophagy in plant root cells. Autophagy 3, 360–362 (2007).
Izumi, M., Wada, S., Makino, A. & Ishida, H. The autophagic degradation of chloroplasts via rubisco-containing bodies is specifically linked to leaf carbon status but not nitrogen status in Arabidopsis. Plant Physiol. 154, 1196–1209 (2010).
Wang, Y., Nishimura, M. T., Zhao, T. & Tang, D. ATG2, an autophagy-related protein, negatively affects powdery mildew resistance and mildew-induced cell death in Arabidopsis. Plant J. 68, 74–87 (2011).
Liu, Y., Xiong, Y. & Bassham, D. C. Autophagy is required for tolerance of drought and salt stress in plants. Autophagy 5, 954–963 (2009).
Aubert, S. et al. Ultrastructural and biochemical characterization of autophagy in higher plant cells subjected to carbon deprivation: control by the supply of mitochondria with respiratory substrates. J. Cell Biol. 133, 1251–1263 (1996).
Strommer, J. The plant ADH gene family. Plant J. 66, 128–142 (2011).
Garabagi, F., Duns, G. & Strommer, J. Selective recruitment of Adh genes for distinct enzymatic functions in Petunia hybrida. Plant Mol. Biol. 58, 283–294 (2005).
Alleva, K. et al. Cloning, functional characterization, and co-expression studies of a novel aquaporin (FaPIP2;1) of strawberry fruit. J. Exp. Bot. 61, 3935–3945 (2010).
Gillaspy, G. E., BenDavid, H. & Gruissem, W. Fruits: a developmental perspective. Plant Cell 5, 1439–1451 (1993).
Ando, K., Carr, K. M. & Grumet, R. Transcriptome analyses of early cucumber fruit growth identifies distinct gene modules associated with phases of development. BMC Genomics 13, 518 (2012).
Ando, K. & Grumet, R. Transcriptional profiling of rapidly growing cucumber fruit by 454-pyrosequencing analysis. J. Am. Soc. Hortic. Sci. 135, 291–302 (2010).
Lemaire-Chamley, M. et al. Changes in transcriptional profiles are associated with early fruit tissue specialization in tomato. Plant Physiol. 139, 750–769 (2005).
Mascarell-Creus, A. et al. An oligo-based microarray offers novel transcriptomic approaches for the analysis of pathogen resistance and fruit quality traits in melon (Cucumis melo L.). BMC Genomics 10, 467 (2009).
Wechter, W. P. et al. Gene expression in developing watermelon fruit. BMC Genomics 9, 275 (2008).
Wang, Y. P. et al. A comparison of sun, ovate, fs8.1 and auxin application on tomato fruit shape and gene expression. Plant Cell Physiol. 60, 1067–1081 (2019).
Gevens, A. J., Ando, K., Lamour, K. H., Grumet, R. & Hausbeck, M. K. A detached cucumber fruit method to screen for resistance to Phytophthora capsici and effect of fruit age on susceptibility to infection. Plant Dis. 90, 1276–1282 (2006).
Liang, D. N. et al. Identification of differentially expressed genes related to aphid resistance in cucumber (Cucumis sativus L.). Sci. Rep. 5, 9645 (2015).
Gao, Y. et al. Transcriptome analysis of genes related to fruit texture in watermelon. Sci. Hortic. 262, 109075 (2020).
Guo, S. G. et al. Comparative transcriptome analysis of cultivated and wild watermelon during fruit development. PLoS ONE 10, e0130267 (2015).
Nakkanong, K., Yang, J. H. & Zhang, M. F. Carotenoid accumulation and carotenogenic gene expression during fruit development in novel interspecific inbred squash lines and their parents. J. Agric. Food Chem. 60, 5936–5944 (2012).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Vaser, R., Sovic, I., Nagarajan, N. & Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119 (2013).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, 351–358 (2005).
Hoede, C. et al. PASTEC: An Automatic Transposable Element Classification Tool. PLoS ONE 9, 106 (2014).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25 (2009).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, 215–225 (2003).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr. Protoc. Hum. Genet. 64, e56 (2003).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics 19, 1–12 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43, e78 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M. & Buell, C. R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7, 327 (2006).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, 121–124 (2004).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 0955–0964 (1997).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, 225–229 (2011).
Koonin, E. V. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 5, 1–28 (2004).
Dimmer, E. C. et al. The UniProt-GO annotation database in 2011. Nucleic Acids Res. 40, 565–570 (2012).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14 (2019).
Mi, H., Muruganujan, A., Ebert, D., Huang, X. & Thomas, P. D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, 419–426 (2019).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287 (2012).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Yang, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics 13, 555–556 (1997).
Han, M. V., Thomas, G. W., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Zwaenepoel, A. & Van de Peer, Y. wgd-simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics 35, 2153–2155 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–550 (2014).
Acknowledgements
This work was supported by grants from the Beijing Municipal Science and Technology Commission (Z191100008619004 and Z191100004019010), the Key Project of “Science and Technology Boost the Economy 2020,” the Special Innovation Ability Construction Fund of the Beijing Academy of Agricultural and Forestry Sciences (20180705 and 20200427), the China Agriculture Research System Project (CARS-23), the Collaborative Innovation Center of Beijing Academy of Agricultural and Forestry Sciences (201915), the Young Investigator Fund of the Beijing Academy of Agricultural and Forestry Sciences (202016), the National Natural Science Foundation of China (31772022), the Natural Science Foundation of Beijing (6182016), and USDA National Institute of Food and Agriculture Specialty Crop Research Initiative (2015-51181-24285).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, L., Wang, Q., Mu, J. et al. The genome and transcriptome analysis of snake gourd provide insights into its evolution and fruit development and ripening. Hortic Res 7, 199 (2020). https://doi.org/10.1038/s41438-020-00423-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-00423-9
