
Abstract
Conservation genetic theory suggests that small and isolated populations should be subject to reduced genetic diversity i.e., heterozygosity and allelic diversity. Our 34 years study of an isolated island population of adders (Vipera berus) in southern Sweden challenges this notion. Despite a lack of gene flow and a yearly mean estimated reproductive adult population size of only 65 adult adders (range 12–171), the population maintains high levels of heterozygosity and allelic diversity similar to that observed in two mainland populations. Even a 14-year major “bottleneck” i.e., a reduction in adult adder numbers, encompassing at least four adder generations, did not result in any reduction in the island adders’ heterozygosity and allelic diversity. Female adders are polyandrous, and fertilisation is non-random, which our empirical data and modelling suggest are underpinning the maintenance of the population’s high level of heterozygosity. Our empirical results and subsequent modelling suggest that the positive genetic effects of polyandry in combination with non-random fertilisation, often overlooked in conservation genetic analyses, deserve greater consideration when predicting long-term survival of small and isolated populations.
Similar content being viewed by others


Introduction
Over the last century, the combination of anthropogenic degradation of natural habitats and the deleterious effects of climate change have resulted in exceedingly isolated and fragmented habitats causing a significant decrease in the population size of numerous organisms and a dramatic loss in global biodiversity (Brook et al. 2008; Pimm et al. 2014; Newbold et al. 2016; Spooner et al. 2018). A severe reduction in population size will frequently result in a major loss of genetic diversity which in turn has been shown to increase the risk of inbreeding depression, resulting in decreased fertility as well as offspring survival and a concomitant increased risk of extinction (Charlesworth and Charlesworth 1987; Roelke et al. 1993; Madsen et al. 1996; Saccheri et al. 1998; Ujvari et al. 2002; Frankham 2005a; Hedrick et al. 2019; Kyriazis et al. 2020).
Population genetic diversity (expected heterozygosity) as well as population genotypic diversity (observed heterozygosity) have, however, been shown not only to be affected by population size but also by the species’ social structure (Parreira and Chikhia 2015). Species living in socially structured populations may maintain high levels of genetic genotypic diversity without invoking sex-biased dispersal or inbreeding avoidance mechanisms (Parreira and Chikhia 2015; Parriera et al. 2020). Another factor, similarly not related to population size, affecting heterozygosity is the genetic effect of assortative mating, a phenomenon recorded in numerous species, as reviewed by Jiang et al. (2013). Positive assortative mating refers to individuals mating with individuals with similar phenotype/genotype to their own, whereas negative assortative mating, also referred to as disassortative mating, refers to individuals mating with individuals with dissimilar phenotype/genotype to their own (Jiang et al. 2013). Positive assortative mating may lead to a decrease in heterozygosity, whereas negative assortative mating may result in increased heterozygosity (Jiang et al. 2013; Hedrick et al. 2018).
Similar to negative assortative mating, polyandry (females mating with multiple partners) can result in increased population genetic diversity (Lewis et al. 2020). Importantly, polyandry may also enhance female fitness (Madsen et al. 1992, 2005; Jennions and Petrie 2000; Tregenza and Wedell 2002; Fisher et al. 2006; Firman and Simmons 2008; Slatyer et al. 2012; Rosengrave et al. 2016; Vuarin et al. 2019; see Supplementary Information for additional references S1–S14, but see Madsen 2011; Ashby and Gupta 2013). The potential positive effects of polyandry have been suggested to arise from enhanced fertilisation success of genetically more divergent males, which has been recorded in numerous organisms (Olsson et al. 1996, 1997; Vacquier 1998; Palumbi 1999; Bonneaud et al. 2006; Palumbi 2009; Gasparini and Pilastro 2011; Evans and Sherman 2013; Løvlie et al. 2013; Gasparini and Evans 2018; Zhang et al. 2020; Madsen et al. 2020) (see Supplementary Information for additional references S15–S36) including humans (Fitzpatrick et al. 2020). This enhanced fertilisation success of genetically more divergent males has been referred to as “cryptic female choice” (Eberhard 1996). As several mechanisms have been suggested to underpin this process, such as sperm motility as well as ova-sperm interactions, reviewed by Firman et al. (2017), we refer to these processes as “non-random fertilisation”.
Based on a long-term study spanning 34 years (1984–2017), we show that a small isolated population of adders (Vipera berus), on the Swedish island of Hallands Väderö has been able to retain high levels of heterozygosity and allelic diversity despite the absence of recent gene flow and being subjected to a major long-term reduction in numbers (population bottleneck) spanning at least four adder generations. In the present study we compare this island’s adder population’s level of heterozygosity and allelic diversity with an adjacent mainland population and a second mainland population subjected to genetic rescue in 1992 that in the subsequent 26 years resulted in a rapid increase in heterozygosity (Madsen et al. 2020, see “Materials and methods” for further information). Importantly, our analyses show no admixture among the populations, which strongly suggests that the high levels of heterozygosity and allelic diversity in the island population were not caused by gene flow from the adjacent mainland. Instead, both our empirical data (Madsen et al. 1992, 1996, 2020; Olsson and Madsen 2001) and the results from our subsequent modelling suggest that polyandry in combination with non-random fertilisation, so far not considered in previous conservation genetic theory/analyses, may underpin how the population has been able to maintain its high level of heterozygosity despite its small population size and long-term isolation.
Materials and methods
Study sites and species
Hallands Väderö, henceforth HV (56.2635oN and 12.3342oE) is an island with an area of 3.1 km2 situated in south-western Sweden (see Supplementary Information Fig. S1). HV has been isolated from the mainland for at least 6000 years and has been a nature reserve since 1958 (Ekstam and Forshed 2010). The long-term isolation from the mainland is indicated by the presence of only one amphibian, the smooth newt (Lissotriton vulgaris) on the island, compared with four abundant amphibians, the common frog (Rana temporaria), moor frog (R. arvalis), common toad (Bufo bufo) and the crested newt (Trirurus cristatus), on the adjacent mainland (Andrén and Nilsson 1979) (and pers.obs.) The second study site, Torekov (56.4236° N, 12.6273° E), is situated on the mainland, 3 km from HV and the third study site, Smygehuk (55.3385° N, 13.3598° E) is situated in the southernmost part of Sweden ~130 km south of HV and Torekov (see Supplementary Information Fig. S1).
The Torekov study site was used to investigate possible gene flow between HV and the adjacent mainland as well as to compare the adders’ heterozygosity and allelic diversity relative to that of HV. The Smygehuk study site was incorporated into the present study as the local adder population was subject to genetic rescue in 1992, which in the subsequent 26 years resulted in a rapid increase in offspring viability, heterozygosity and a concomitant increase in adder numbers (Madsen et al. 2020). In comparison, the Smygehuk population provides a reference for a population with high levels of heterozygosity and allelic diversity.
Adders are small venomous snakes widely distributed through Europe and Asia, where females normally grow to 65 cm snout-vent length and males to 55 cm (Arnold and Burton 1978). Our previous studies show that adder generation time is 3 years (Madsen and Shine 1994) and that female adders are polyandrous, mating with on average 3.7 different males during their spring reproductive period (Madsen et al. 1992). Importantly, our previous studies also show that fertilisation is non-random (Madsen et al. 1992, 1996, 2020; Olsson and Madsen 2001). Female adders are viviparous and, in southern Sweden, give birth to a mean brood size of 9.2 of which, on average, 2 survive to a reproductive age (Madsen and Shine 1994).
Field work and estimates of annual abundance
Each year the island of HV was visited on two separate occasions during cold but sunny days in March to early April. Sampling effort was consistent across years and the same standardised routes covering optimal adder habitats, i.e. the edges of thick stands of blackthorn (Prunus spinosa) and juniper (Juniperus communis) bushes and the adjacent meadows were used throughout the study. Although the island has a total area of 3.1 km2 based on our long-term field work with adders, spanning more than 40 years (Madsen et al. 2020, 2022), in early spring only a fraction (about 10%) of the island is an optimal adder habitat. During this time of the year the snakes are confined close to the edges of the dense bushes and the adjacent grassy meadows. Moreover, due to the low ambient temperatures in early spring, the adult male adders are exposed to the sun for most of the day, greatly facilitating them being captured. We are therefore convinced that each year of the study most of the adult males were indeed captured. Time between visits ranged from 7 to 14 days. From 1984 to 2017, 357 adult males were individually marked by ventral scale clipping while we also recorded body mass and body length. In late February to early March, adult male adders emerge from hibernation at low ambient temperatures to bask in sun exposed microhabitats, which facilitates their capture (Madsen et al. 2020). Our estimates of HV adder numbers are therefore based on adult males. The adult sex ratio of adders is close to 1:1 (Madsen and Shine 1994). However, as females on average reproduce only every second year, the mean operational sex ratio is male biased. Consequently, the mean proportion of males to reproductive females is 0.667 males vs. 0.333 reproductive females (Madsen and Shine 1993; Bauwens and Claus 2019). We therefore calculated the reproductive adder population size as: total number of reproductive adders = 1.5 × estimated number of adult males.
The POPAN-formulation (Schwarz and Arnason 1996) of the Cormack-Jolly-Seber capture-recapture method (Lebreton et al. 1992; Cooch and White 2015) was used to estimate yearly abundance of adult males. The original daily capture data were collapsed into yearly intervals, such that the encounter history file included for each individual snake one 1/0 (= captured/not-captured) entry per year of study (1984–2017). Models were built that included different combinations of either constant or time-dependent (i.e., among year) probabilities of capture, survival and entrance into the population. We used an information theoretic approach to rank the models according to the sample-size adjusted Akaike’s Information Criterion (Burnham and Anderson 2002; Anderson 2008). All models were fitted using maximum-likelihood methods implemented in programme MARK (Cooch and White 2015; White and Burnham 1999) through the RMark interface package (Laake and Rexstad 2015) in R v4.0.2 (R Core Team 2014). All modelling specifications, including adjustments for different interval length between successive years, follow procedures outlined in Laake and Rexstad (2015). For further information see Supplementary Information in Table S1.
Genetic samples
In both 1996 and 2017, 18 adders were blood sampled on HV. In 2019 and 2020 a total of 12 samples were obtained from the adjacent mainland in Torekov. In both 2005 and 2017, 18 adders were obtained from the third mainland population in Smygehuk. The blood samples were stored in 70% ethanol and DNA was isolated by phenol–chloroform extraction. The samples’ sizes fulfil the numbers to be used based on single nucleotide polymorphisms (SNPs) to make robust estimates of genetic diversity (Nazareno et al 2017).
Studies in other species have failed to detect evidence of non-random fertilisation (Fleishman and Sakaluk 2004; Denk et al. 2005; Simmons et al. 2006; Bos et al. 2009; Ala-Honkola et al. 2011; Mongue et al. 2014; Huang and Pemberton 2021). The discrepancy in the results from these studies compared with those mentioned earlier in the introduction that did, may to a certain extent be due to the molecular markers used. For example, in mouse lemurs (Microcebus murinus) the effect of inbreeding avoidance along with disassortative mate choice was observed at MHC class II at the DRB gene, but not at the DQB gene (Huchard et al. 2013). In the present study we therefore used SNP markers that enable genome-wide analyses of genetic diversity (e.g. Morin et al. 2004; Miller et al. 2014).
Sequencing for SNP genotyping was performed by DArTseqTM (DArT Pty Ltd, Canberra), after complexity reduction using a combination of restriction enzymes. For fragment size selection and next generation sequencing details, see Kilian et al. (2012) and Georges et al. (2018). To achieve the most appropriate complexity reduction (the fraction of the genome represented, controlling average read depth and number of polymorphic loci), four combinations of restriction enzymes (Pstl enzyme combined with either Hpall, Sphl, Nspl or Msel) were evaluated and the restriction enzyme combination of Pstl (recognition sequence 5’-CTGCA|G-3’) and Sphl (5’-GCATG|C-3’) was selected.
Only fragments generated by the Psti-Sphl double digest were effectively amplified in 30 rounds of polymerase chain reaction (PCR). Amplifications consisted of an initial denaturation step of 94 °C for 1 min, followed by 30 cycles of PCR with the following temperature profile: denaturation at 94 °C for 20 s, annealing at 58 °C for 30 s and extension at 72 °C for 45 s, with an additional final extension at 72 °C for 7 min. After PCR, equimolar amounts of amplification products from each sample were pooled and applied to cBot (lllumina) bridge PCR for sequencing on the lllumina Hiseq 2500. The sequencing (single end) was run for 77 cycles.
Sequences generated from each lane were processed using proprietary DArT Pty Ltd analytical pipelines as described by Georges et al. (2018). In particular, one third of samples were processed twice from DNA, using independent adaptors, to allelic calls as technical replicates, and scoring consistency (repeatability) was used as the main selection criterion for high quality/low error rate markers. The resultant data set contained the SNP genotypes and various associated metadata of which CloneiD (unique identity of the sequence tag for a locus), repAvg (proportion of technical replicate assay pairs for which the marker score is identical), CallRate (proportion of individuals scored at a particular locus) and SnpPosition (position in the sequence tag at which the defined SNP variant base occurs) used in our analyses.
The SNP data and associated metadata were read into a genlight object (Jombart 2008) to facilitate processing with package dartR (Gruber et al. 2018). Only loci with 99% or greater repeatability (repAvg) were chosen for subsequent analysis. Further filtering was undertaken on the basis of call rate (95%). Finally, we filtered out secondary SNPs where they occurred in a single sequenced tag, retaining only one SNP at random. This additional filtering resulted in 7504 SNP markers for the 84 adders, and we regard the data remaining after filtering to be highly reliable.
Visualisation of population genetic structure
Genetic similarity among individuals and populations was visualised using ordination principal components analysis or PCA (Jolliffe 2002) as implemented in the gl.pcoa and gl.pcoa.plot functions of dartR. A scree plot of eigenvalues guided the number of informative axes to examine (Jolliffe 2002), taken in the context of the average percentage variation explained by the original variables (using the gl.pcoa.scree function in dartR).
Genetic diversity
We estimated two complementary measures of genetic diversity, expected heterozygosity and allelic diversity. We compared the expected heterozygosity for each of the three populations based on allele frequencies in the gl.report.heterozygosity function of dartR66. Pairwise comparisons of expected heterozygosity among the three populations were analysed by the gl.test.heterozygosity function in dartR (pairwise significance for each comparison estimated by re-randomisation with 20,000 replicates). Average allelic diversity is independent from estimates of allelic frequencies and therefore an estimator of allelic diversity within a population, though potentially biased if not corrected for sample sizes due to missed rare alleles (Leberg 2002). Hence, we corrected for differences in sample size, by down sampling estimates to the lowest number of samples (i.e. Torekov N = 12), by randomly drawing 50 times 12 individuals from populations and then calculating allelic diversity for each subsample. Calculations were performed using the gl.report.diversity function in dartR (Gruber et al. 2018). Typically, heterozygosity estimates based on both micro satellites and SNPs show that micro satellite estimates are approximately twice as high as those obtained using SNPs (Miller et al. 2014; Zimmerman et al. 2020).
Effective population size
The effective population size of the HV adders in 1996 and 2017 was estimated based on molecular linkage disequilibrium estimated using the bias-corrected method (Hill 1981; Waples 2006) (as implemented in NeEstimator v2.1, Do et al. 2014). Confidence intervals were based on the Jacknife methods, on individuals as implemented in NeEstimator.
To estimate the expected loss of heterozygosity during the 14-year bottleneck (see below) we created a synthetic population with an initial heterozygosity of 0.5 for 7504 loci (please note the initial amount of heterozygosity and the number of loci do not matter, as we were interested in the relative decline compared to the starting population) and simulated the heterozygosity for our estimated Ne = 21 (see below), with 10,000 repeats with non-overlapping generations for 4 and 5 generations (generation time 3 years, Madsen and Shine 1994) and extrapolated for 4.7 generations, which was determined as the length of the bottleneck (14 years/3). To make sure our estimate of Ne is not influencing our conclusion that a significant loss during the bottleneck should have happened, we assumed that our estimate of Ne = 21 was biased 50% low (which is the maximal bias found by Waples et al. (2014) for a selection of 22 species), we re-ran our simulation for an estimate of Ne = 42. We used function gl.sim.ind as implemented in the dartR package, which generates future generations based on the allele frequencies of individuals of the current population (see Supplementary Information for the simulation code).
Modelling the temporal effects of polyandry and non-random fertilisation on population genetic diversity
The aim of the model is to show how polyandry and non-random fertilisation, whereby females favour sperm of genetically dissimilar mates (Fig. 1), can assist in maintaining long-term genetic diversity in small animal populations. The model is not meant to predict exact changes in heterozygosity in a specific adder population, but rather meant to illustrate the contribution that polyandry in combination with non-random fertilisation can have on promoting and maintaining long-term genetic diversity in small populations. The model is thus parsimonious and only loosely parameterised, only roughly resembling a small population of adders.

Non-random high fertilisation is based on empirical results from Olsson et al. (1996).
The model assumes:
-
(1)
Random mating (Madsen et al. 1992).
-
(2)
Reproduction is seasonal and takes place every year (Madsen and Shine 1993, 1994; Bauwens and Claus 2019).
-
(3)
Proportion of adult males is 0.667 (Madsen and Shine 1993; Bauwens and Claus 2019).
-
(4)
Each year all females copulate with four random males, following findings of on average 3.7 different males per female (Madsen et al. 1992) and give birth to 9 offspring of which two survive till reproductive age (Madsen and Shine 1994). While in reality reproductive age is reached at 3 years of age on average (Madsen and Shine 1994), for simplicity these 6 offspring enter the reproducing population after 1 year.
-
(5)
The chance of males fertilising eggs declines with their genetic similarity to the female (Olsson et al. 1996).
-
(6)
Mortality is random amongst adult individuals and is strictly density dependent (i.e., it is tuned to keep the total reproductive population size at 40 individuals).
-
(7)
The model includes three levels of initial heterozygosity (i.e., low = 0.05, medium = 0.20 and high = 0.40). For further information on the model, including model code, see supplementary information.
Results
Population dynamics
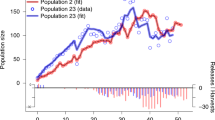
The most parsimonious model to estimate adult male annual abundance assumed constant among year capture rates (see Supplementary Information Table S1) which resulted in a significant relationship between estimated adult male number and number captured (r2 = 0.98, p < 0.0001, n = 22). The mean annual number of adult males captured ranged from 4 in 1997 to 61 in 1984 (mean 23), annual adult male population size was estimated to range from 8 in 1997 to 114 in 1984 (mean 43) and estimated mean annual reproductive population size was estimated to range from 12 in 1997 to 171 in 1984 (mean 65, Fig. 2). Consequently, annual captures showed a substantial among-year variation. This variation in numbers was due to the number of adult males captured from 1984 to 1996 and in 2017 was significantly higher than during the 14 years from 1997 to 2010 (F1,21 = 31.1, p < 0.0001, mean 33.1 vs. 10.4, respectively).

The horizontal blue line depicts the duration of the bottleneck.
Genetic diversity and structure
Expected heterozygosity and allelic diversity analyses showed no significant difference among the 1996 and 2017 HV samples (Table 1). The expected heterozygosity of the HV adders was significantly higher than that recorded in the Torekov population on the adjacent mainland, whereas no difference in allelic diversity was observed between the two populations (Table 1). Finally, the expected heterozygosity and the allelic diversity of the HV population was indistinguishable from the second mainland Smygehuk population that had been subjected to genetic rescue (Madsen et al. 2020; Table 2). No differences in allelic diversity across the five samples from the three populations were observed. Note that as SNPs are bi-allelic the maximum allelic diversity is 2.0.
The population genetic structure among the three populations was distinct with no intermediate individuals, strongly suggesting no admixture, i.e. a lack of gene flow among the three populations (Fig. 3). Analyses of the fixation index (Fst), a measure of population differentiation in genetic structure, revealed significant divergence among the three populations (all pairwise comparisons p < 0.0001, based on 20,000 permutations, Table 2). Together, these results strongly suggest no recent or contemporary gene flow between HV and the two mainland populations.

Each data point is 1 individual.
Effective population size
Linkage disequilibrium analyses suggested a two-fold increase in Ne from 1996 to 2017 (from 74.8 to 157.9) but based on capture-mark-recapture results only a slight increase in estimated population size was observed over this period (from 112 to 138; Table 3).
Based on the body length of adders captured in 1996 and 2017 the population consisted of four major age cohorts (Madsen and Shine 1994). As adder generation time is 3 years (Madsen and Shine 1994), the average number of age cohorts/generation ratio was 1.3, which according to Waples et al. (2014) would generate a low bias caused by the effect of overlapping generations in the two observed effective population size estimates.
The harmonic mean of the estimated reproductive population size during the 14-year bottleneck was 25 (see Supplementary Information Table S2) and the harmonic mean Ne/N ratio for 1996 and 2017 was 0.84 (calculated from Table 1), similar to the mean Ne observed in 16 vertebrates presented in Table S1 in Waples et al. (2014).
This suggests a mean bottleneck Ne of 21 (i.e. 25 × 0.84). We estimated the expected reduction in heterozygosity of the bottleneck using the method described by Frankham et al. (2010), i.e. [1−1/(2Ne)]t, where t depicts number of generations. As generation time in adders is 3 years, the 14-year bottleneck covered 4.7 generations (i.e. 14/3) and the expected reduction in heterozygosity was 0.0893, from 0.239 in 1996 to 0.217 in 2017. However, despite this expected 9% decline in heterozygosity, no reduction in heterozygosity was observed (Table 1).
We subsequently simulated the loss of heterozygosity caused by the bottleneck with an Ne of 21 for 4.7 generations (based on 10,000 simulations, see methods and Supplementary Information for details and simulation code). The model results were similar to the theoretical expected loss of heterozygosity, i.e. 0.0886 vs. 0.0893, respectively. The 95% confidence interval ranged from 0.0860 to 0.0904 and did not include zero. Importantly, to ensure that our result was not based on a potential bias in our estimate of Ne, we re-ran the simulation for an Ne = 42 (based on the maximum bias of 50%, estimated from Fig. 6 (excluding mosquitoes) in Waples et al. 2014), which resulted in a mean loss of heterozygosity of 0.0941 (95% confidence interval 0.0931–0.0949; see Supplementary Information Fig. S2). In summary, all three approaches yielded an expected reduction of heterozygosity of ~9% while the empirical data showed no such decline.
Modelling the effects of polyandry and random vs. non-random fertilisation
At all three levels of initial heterozygosity, our modelling showed that polyandry in combination with random fertilisation resulted in a temporal decrease of heterozygosity (Fig. 4a–c). At low and intermediate levels of initial heterozygosity both non-random intermediate fertilisation and non-random high levels of fertilisation showed an initial increase in heterozygosity and thereafter a stable level (Fig. 4d, e, g, h). Finally, at high levels of initial heterozygosity and non-random intermediate fertilisation there was a tendency for reduction in heterozygosity (Fig. 4f). However, at non-random high levels of fertilisation the level of heterozygosity remained stable (Fig. 4i).

Each panel depicts the median (red line), 25–75 percentile range (dark pink area) and min–max range (light pink area) across 100 simulations. Panels depict different levels of fertilisation. a–c Random fertilisation with heterozygosity ranging from low to high, d, e non-random intermediate fertilisation with heterozygosity ranging from low to high, and g–i non-random high fertilisation with heterozygosity ranging from low to high.
Discussion
The importance of genetic diversity and genotypic diversity on population viability has been documented in numerous studies (Roelke et al. 1993; Lacy 1997; Reed et al. 2003; Frankham 2005b; Foerster et al. 2007; Markert et al. 2010; Allentoft and O’Brien 2010; Madsen et al. 2020; Scott et al. 2020), but see Teizeria and Huber (2021), and, as mentioned above, reduced genetic diversity may increase the risk of populations becoming subjected to inbreeding depression and concomitant increased risk of extinction (Charlesworth and Charlesworth 1987; Roelke et al. 1993; Madsen et al. 1996; Saccheri et al. 1998; Ujvari et al. 2002; Frankham 2005a; Hedrick et al. 2019; Kyriazis et al. 2020). Therefore, how small population size and low population genetic diversity will impact on long-term population viability has been and continues to be a central question in conservation genetics (Mace and Lande 1991; Ellstrand and Elam 1993; Lynch et al. 1995). Conservation genetics studies have suggested that an effective population size (Ne) of 1000 is required to maintain long-term population viability/survival (Frankham et al. 2014) and therefore estimates of Ne have been advocated to be imperative in predicting estimates of long-term population viability (Frankham et al. 2014).
Our two estimates of Ne of the HV adder population were based on molecular linkage disequilibrium (Hill 1981; Waples 2006). The first estimate was based on samples collected in 1996, prior to a 14-year decline in adder numbers, and the second estimate from samples collected in 2017 when adder numbers had recovered to numbers similar to that recorded in 1996. Our estimates of Ne in 1996 was about half that recorded in 2017 (74.9 and 157.9), whereas our estimates on the population’s expected heterozygosity and allelic diversity in 1996 and 2017 were identical and well below clearly below the 1000 suggested to be required to maintain long-term population viability/survival by Frankham et al. (2014).
As mentioned above, population genetic modelling in Parreira and Chikhia (2015) showed how species living in social groups were able to maintain high genotypic diversity without invoking sex-biased dispersal or inbreeding avoidance mechanisms. Adders, however, do not live in social groups and females mate with any male able to access the female (Madsen et al. 1992). Moreover, the modelling by Parreira and Chikhia (2015) and by Parriera et al. (2020) did not include active inbreeding avoidance mechanisms whereas our modelling estimated the genetic effects of non-random fertilisation based on male–female genetic similarity i.e., inbreeding avoidance. Our modelling showed that random fertilisation resulted in gradual temporal reduction in heterozygosity, whereas non-random fertilisation promoted and maintained long-term levels of heterozygosity. The results of the model are consistent with the empirical data, which shows that despite its small population size and lack of gene flow, the HV population has been able to maintain a higher level of heterozygosity and allelic diversity than the Torekov mainland adders, where adders commonly occur along a 15 km coastline (Madsen pers. obs.). At the same time the level of heterozygosity and allelic diversity at HV is similar to that of the Smygehuk population, which was subject to genetic rescue resulting in a significantly increased heterozygosity, from 0.170 prior to the rescue to a mean of 0.242 after the rescue (Madsen et al. 2020). High genetic diversity of small populations has also been recorded in other taxa such as blue whale (Balaenoptera musculus) (Torres-Florez et al. 2014), white-tailed eagle (Haliaeetus albicilla) (Hailer et al. 2006), copper redhorse (Moxostoma hubbsi) (Lippé et al. 2006) and Drosophila melanogaster (Schou et al. 2017). In blue whale, white-tailed eagle and copper redhorse the maintenance of genetic diversity has been suggested to be caused by long generation time. As the generation time in adders is 3 years (Madsen and Shine 1994), we find it unlikely that this demographic parameter underpins the island’s adder population’s high level of heterozygosity and allelic diversity genetic diversity. In Drosophila melanogaster Schou et al. (2017) suggested that maintenance of genetic diversity was caused by “associative overdominance, driven by disfavoured homozygosity of recessive deleterious alleles”. The latter hence suggests a somewhat similar scenario to that provided in the present study.
A major temporal reduction in population size, i.e., a population bottleneck, has been shown to reduce genetic diversity and hence reduce a population’s evolutionary potential (Keller et al. 1994; Frankham et al. 1999; Kuo and Janzen 2004; Price and Hadfield 2014; Sonsthagen et al. 2017). From 1996 to 1997 adder numbers on the island dropped dramatically and remained low until 2010. Major declines in prey availability have been shown to result in emaciation, reduced survival, and concomitant population reduction in water python (Liasis fuscus) (Ujvari et al. 2016). However, the HV adders captured during the 14-year bottleneck were not emaciated, strongly suggesting that the bottleneck was not caused by a reduction of field voles (Microtus agrestis), which constitutes the main prey of adult adders on the island (pers. obs). Moreover, no other apparent habitat changes occurred on HV potentially explaining the bottleneck. While the cause of the bottleneck is thus still unclear, our analyses suggest that genetic drift should have resulted in a 9% reduction in heterozygosity. However, our empirical data did not show any reduction of heterozygosity from 1996 to 2017. We therefore suggest that the adders’ ability to maintain a high level of heterozygosity was due to the positive genetic effects of polyandry in combination with non-random fertilisation. This explanation is also supported by the outcome of our modelling of the effects of polyandry and random vs. non-random fertilisation.
Importantly, our modelling also showed that if a population is subjected to extremely low levels of heterozygosity, polyandry and non-random fertilisation, this will not enhance heterozygosity to sufficient levels to maintain long-term viability. Genetically eroded populations like the Smygehuk adder population, initially suffering from severe inbreeding depression (Madsen et al. 1996) may therefore require genetic rescue to increase their genetic diversity to a level where polyandry and non-random fertilisation may maintain long-term genetic viability (Madsen et al. 2020).
Presently, numerous species are threatened due to low population numbers, concomitant reduced genetic diversity and the negative effects of stochastic processes such as genetic drift (Lande 1976). Climate change is predicted to increase the frequency of extreme environmental perturbations (Easterling et al. 2000; Parmesan et al. 2000) and the associated negative effects on population demography resulting in an increased risk of extinction (Thibault and Brown 2008; Ujvari et al. 2016). The positive effects on heterozygosity of polyandry in combination with non-random fertilisation observed in adders and numerous other taxa (see references in the introduction), may therefore provide a glimmer of hope that some of the presently threatened taxa, expressing similar genetic diversity to that observed in adders on Hallands Väderö, may therefore be able to survive the ongoing 6th extinction (Ceballosa et al. 2017). This notion warrants further in-depth studies into the prevalence and extent of polyandry and non-random fertilisation in other organisms.
Conclusion
The positive effects of polyandry and non-random fertilisation on population genetic diversity have been largely ignored in conservation genetics theory/analyses. Our study suggests that the positive genetic effects of polyandry in combination with non-random fertilisation should be considered when predicting long-term survival of small populations.
Data availability
SNP data used in the analyses, unfiltered and filtered as per Methods, have been deposited on DRYAD (https://doi.org/10.5061/dryad.73n5tb31c).
References
Ala-Honkola O, Manier MK, Lüpold S, Pitnick S (2011) No evidence for postcopulatory inbreeding avoidance in Drosophila melanogaster. Evolution 65:2699–2705
Allentoft ME, O’Brien J (2010) Global amphibian declines, loss of genetic diversity fitness, a review. Diversity 2:47–71
Anderson DR (2008) Model based inference in the life sciences—a primer on evidence. Springer, New York
Andrén C, Nilsson G (1979) Reptiler och amfibier på Hallands Väderö. Fauna och Flora 5:193–201
Arnold EN, Burton JA (1978) A field guide to the reptiles and amphibians of Britain Europe. Collins, London
Ashby B, Gupta S (2013) Sexually transmitted infections in polygamous mating systems. Philos Trans R Soc B 368:20120048
Bauwens D, Claus K (2019) Intermittent reproduction, mortality patterns and lifetime breeding frequency of females in a population of the adder (Vipera berus). PeerJ 7:e6912
Bonneaud C, Chastel O, Federici P, Westerdahl H, Sorci G (2006) Complex Mhc-based mate choice in a wild passerine. Proc R Soc B 273:1111–1116
Bos DH, Williams RN, Gopurenko D, Bulut Z, DeWoody JA (2009) Condition dependent mate choice and reproductive disadvantages for MHC-divergent male tiger salamanders. Mol Ecol 15:3307–3315
Brook BW, Sodhi NS, Bradshaw CJA (2008) Synergies among extinction drivers under global change. Trends Ecol Evol 23:453–460
Burnham KP, Anderson DR (2002) Model selection and multimodel inference: a practical information-theoretic approach. Springer, New York
Ceballosa G, Ehrlich PR, Dirzob R (2017) Biological annihilation via the ongoing sixth mass extinction signaled by vertebrate population losses and declines. Proc Natl Acad Sci USA 114:E6089–E6096
Charlesworth D, Charlesworth B (1987) Inbreeding depression its evolutionary consequences. Ann Rev Ecol Syst 18:237–268
Cooch EG, White GC (2015) Program Mark—a gentle introduction. http://www.phidot.org/software/mark/docs/book/
Denk A, Holzman A, Peters A, Vermierssen ELM, Kempenaers B (2005) Paternity in mallards, effects of sperm quality female sperm selection for inbreeding avoidance. Behav Ecol 16:825–833
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2, re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Easterling DR, Meehl GA, Parmesan C, Changnon SA, Karl TR, Mearns LO (2000) Climate extremes, observations, modelling, and impacts. Science 289:2068–2074
Eberhard WG (1996) Female control, sexual selection by cryptic female choice. Princeton University Press, Princeton, NJ
Ekstam U, Forshed N (2010) Hallsands Väderö naturen och historien. Ljungbergs tryckeri, Klippan, Sweden
Ellstrand NC, Elam DR (1993) Population genetic consequences of small populations size, implications for plant conservation. Ann Rev Ecol Syst 24:217–242
Evans JP, Sherman CDH (2013) Sexual selection the evolution of egg-sperm interactions in broadcast-spawning invertebrates. Biol Bull 224:166–183
Fleishman RR, Sakaluk SK (2004) No direct or indirect benefits to cryptic female choice in house crickets (Acheta domestica). Behav Ecol 15:793–798
Firman RC, Simmons LW (2008) Polyandry facilitates postcopulatory inbreeding avoidance in house mice. Evolution 62:603–611
Firman RC, Gasparini C, Manier MK, Pizzari T (2017) Postmating female control, 20 years of cryptic female choice. Trends Ecol Evol 32:368–382
Fisher DO, Double MC, Blomberg SP, Jennions MD, Cockburn A (2006) Post-mating sexual selection increases lifetime fitness of polyandrous females in the wild. Nature 444:89–92
Fitzpatrick JL, Willis C, Devigili A, Young A, Carroll M, Hunter HR et al. (2020) Chemical signals from eggs facilitate cryptic female choice in humans. Proc R Soc B 287:20200805
Foerster K, Coulson T, Sheldon BC, Pemberton JM, Clutton-Brock T, Kruuk LEB (2007) Sexually antagonistic genetic variation for fitness in red deer. Nature 447:1107–1111
Frankham R, Lees K, Montgomery ME, Engl PR, Lowe EH, Briscoe DA (1999) Do population size bottlenecks reduce evolutionary potential? Anim Conserv 2:255–260
Frankham R (2005a) Genetics and extinction. Biol Conserv 126:131–140
Frankham R (2005b) Stress adaptation in conservation genetics. Evol Biol 18:750–755
Frankham R, Ballou JD, Briscoe DA (2010) Introduction to conservation genetics. Cambridge University Press, Cambridge, UK
Frankham R, Bradshaw CJA, Brook BB (2014) Genetics in conservation management, revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol Conserv 170:56–63
Gasparini C, Pilastro A (2011) Cryptic female preference for genetically unrelated males is mediated by ovarian fluid in the guppy. Proc R Soc B 278:2495–2501
Gasparini C, Evans JP (2018) Female control over multiple matings increases the opportunity for postcopulatory sexual selection. Proc R Soc B 285:20181505
Georges A, Gruber B, Pauly GB, White DF, Adams M, Young MJ et al. (2018) Genomewide SNP markers breathe new life into phylogeography and species delimitation for the problematic short-necked turtles (Chelidae, Emydura) of eastern Australia. Mol Ecol 27:5195–5213
Gruber B, Unmack P, Berry OF, Georges A (2018) DARTR, an R package to facilitate analysis of SNP data from reduced representation genome sequencing. Mol Ecol Res 18:691–699
Hailer F, Heler B, Folkestad AO, Ganusevich SA, Garstad S, Hauff P et al. (2006) Bottlenecked but long-lived, high genetic in white-tailed eagles upon recovery from population decline. Biol Lett 22:316–319
Hill WG (1981) Estimation of effective population size from data on linkage disequilibrium. Genet Res 38:209–216
Hedrick PW, Tuttle EM, Rusty A, Gonser RA (2018) Negative-assortative mating in the white-throated sparrow. J Heredity 109:223–231
Hedrick PW, Robinson JA, Peterson RO, Vucetich JA (2019) Genetics and extinction the example of Isle Royale wolves. Anim Conserv 22:302–309
Huang W, Pemberton JM (2021) Within-trio tests provide little support for post-copulatory selection on MHC haplotypes in a free-living population. Proc R Soc B 288:20202862
Huchard E, Baniel A, Schliehe-Diecks S, Kappeler PM (2013) MHC-disassortative mate choice and inbreeding avoidance in a solitary primate. Mol Ecol 22:4071–4086
Jennions MD, Petrie M (2000) Why do females mate multiply? A review of the genetic benefits. Biol Rev 75:21–64
Jiang Y, Bolnick DI, Kirkpatrick M (2013) Assortative mating in animals. Am Nat 181:E125–138
Jolliffe I (2002) Principal component analysis. Springer International Publishing, New York City, USA
Jombart T (2008) adegenet, a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Keller LF, Arcese P, Smith JNM, Hochaka WM, Stearns SC (1994) Selection against inbred song sparrows during a natural population bottleneck. Nature 373:356–357
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H et al. (2012) Diversity arrays technology, a generic genome profiling technology on open platforms. Methods Mol Biol 888:67–89
Kuo C-H, Janzen FJ (2004) Genetic effects of a persistent bottleneck on a natural population of ornate box turtles (Terrapene ornata). Conserv. Gen 5:425–437
Kyriazis CC, Wayne RK, Lohmueller KE (2020) Strongly deleterious mutations are a primary determinant of extinction risk due to inbreeding depression. Evol Lett 5:33–47
Laake J, Rexstad E (2015) RMark—an alternative approach to building linear models in MARK. In: Cooch EG, White GC (eds) Program Mark—a gentle introduction. eBook, Appendix C 1- 109. http://www.phidot.org/software/mark/docs/book/
Lacy RC (1997) Importance of genetic variation to the viability of mammalian populations. J Mamm 78:320–335
Lande R (1976) Natural selection from genetic drift in phenotypic evolution. Evolution 30:314–334
Leberg PL (2002) Estimating allelic richness: effects of sample size and bottlenecks. Mol Ecol 11:2445–2449
Lebreton JD, Burnham KP, Clobert J, Anderson DR (1992) Modelling survival and testing biological hypotheses using marked animals: a unified approach with case studies. Ecol Monogr 62:67–118
Lewis R, Pointer MD, Friend LA, Vasudeva R, Bemrose J, Sutter A et al. (2020) Polyandry provides reproductive genetic benefits in colonising populations. Ecol Evol 10:10851–10857
Lippé C, Dumont P, Bernatchez L (2006) High genetic diversity and no inbreeding in the endangered copper redhorse (Moxostoma hubbsi (Catostimidae, Pisces), the positive effect of a long generation time. Mol Ecol 15:1769–1780
Løvlie H, Gillingham MAF, Worley K, Pizzari T, Richardson DS (2013) Cryptic female choice favours sperm from major histocompatibility complex-dissimilar males. Proc R Soc B 280:20131296
Lynch M, Conery J, Burger R (1995) Mutation accumulation and the extinction of small populations. Am Nat 146:489–518
Mace GM, Lande R (1991) Assessing extinction threats-toward a re-evaluation of IUCN threatened species categories. Conserv Biol 5:148–157
Madsen T, Shine R, Loman J, Håkansson T (1992) Why do female adders copulate so frequently? Nature 335:440–441
Madsen T, Shine R (1993) Temporal variability in sexual selection on reproductive tactics and body size in male snakes. Am Nat 141:167–171
Madsen T, Shine R (1994) Costs of reproduction influence the evolution of sexual size dimorphism in snakes. Evolution 48:1389–1397
Madsen T, Stille B, Shine R (1996) Inbreeding depression in an isolated population of adders (Vipera berus). Biol Conserv 75:113–118
Madsen T, Ujvari B, Olsson M, Shine R (2005) Paternal alleles enhance female reproductive success in tropical pythons. Mol Ecol 14:1783–1787
Madsen T (2011) Cost of multiple matings in female adders (Vipera berus). Evolution 65:1823–1825
Madsen T, Loman J, Anderberg H, Anderberg L, Georges A, Ujvari B (2020) Genetic rescue restores longterm viability of an isolated population of adders (Vipera berus). Curr Biol 30:R1283–R1300
Madsen T, Stille B, Ujvari B, Bauwens D, Endler JA (2022) Negative frequency-dependent selection on polymorphic color morphs in adders. Curr Biol 32:1–4
Markert JA, Champlin DM, Gutjar-Gobell R, Grear JS, Kuhn A, McFreevy Jr TJ et al. (2010) Population genetic diversity and fitness in multiple environments. BMC Evol Biol 10:205
Miller JM, Malenfant RM, David P, Davis CS, Poissant J, Hogg J et al. (2014) Estimating genome-wide heterozygosity: effects of demographic history and marker type. Heredity 112:240–247
Mongue AJ, Ahmed MZ, Tsai MV, de Roode JC (2014) Testing for cryptic female choice in monarch butterflies. Behav Ecol 26:386–395
Morin PA, Luikart G, Wayne RK, and the SNP Workshop Group* (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evol 19:208–216
Nazareno AG, Bemmels JB, Dick CW, Lohmann LG (2017) Minimum sample sizes for population genomics: an empirical study from an Amazonian plant species. Mol Ecol Res 17:1136–1147
Newbold T, Hudson LN, Arnell A, Contu S, De Palma A, Ferrier S et al. (2016) Has land use pushed terrestrial biodiversity beyond the planetary boundary? A global assessment. Science 353:288–291
Olsson M, Shine R, Madsen T, Gullberg A, Tegelström H (1996) Sperm selection by females. Nature 383:585
Olsson M, Shine R, Madsen T, Gullberg A, Tegelström H (1997) Sperm choice by females. Trends Ecol Evol 12:445–446
Olsson M, Madsen T (2001) Promiscuity in sand lizards (Lacerta agilis) and adders (Vipera berus): causes and consequences. J Hered 92:190–197
Palumbi SR (1999) All males are not created equal, fertility differences depend on gamete recognition polymorphisms in sea urchins. Proc Natl Acad Sci USA 96:12632–12637
Palumbi SR (2009) Speciation the evolution of gamete recognition genes, pattern and process. Heredity 102:66–76
Parreira BR, Chikhia L (2015) On some genetic consequences of social structure, mating systems, dispersal, and sampling. Proc Natl Acad Sci USA 112:E3318–E3326
Parriera BR, Quéméré E, Vanpé C, Carvalho I, Chikhia L (2020) Genetic consequences of social structure in the golden-crowned sifaka. Heredity 125:328–329
Parmesan C, Root TT, Willig MR (2000) Impacts of extreme weather and climate on terrestrial biota. Bull Am Meterorol Soc 81:443–450
Pimm SL, Jenkins CN, Abell R, Brooks TM, Gittleman JL, Joppa LN, Raven PH, Roberts CM, Sexton JO (2014) The biodiversity of species and their rates of extinction, distribution, and protection. Science 344:1246752
Price MR, Hadfield MG (2014) Population genetics and the effects of a severe bottleneck in an ex situ population of critically endangered Hawaiian tree snails. PLoS ONE 9:e114377
R Core Team (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Reed DH, O’Grady JJ, Brook BB, Balou JB, Frankham R (2003) Estimations of minimum viable population sizes and factors influencing those estimates. Biol Conserv 113:23–34
Roelke ME, Martenson JS, O’Brien J (1993) The consequences of demographic reduction and genetic depletion in the endangered Florida panther. Curr Biol 3:340–350
Rosengrave P, Montgomerie R, Gemmell N (2016) Cryptic female choice enhances fertilization success and embryo survival in chinook salmon. Proc R Soc B 283:20160001
Saccheri I, Kuussaari M, Kankare M, Vikman P, Fortelius W, Hanski I (1998) Inbreeding and extinction in a butterfly metapopulation. Nature 392:491–494
Schou MF, Loeschke V, Bechsgaard J, Schlötterer C, Kristensen TN (2017) Unexpected high genetic diversity in small populations suggests maintenance by associative overdominance. Mol Ecol 26:6510–6523
Schwarz CJ, Arnason AN (1996) A general methodology for the analysis of open-model capture recapture experiments. Biometrics 52:860–873
Scott PA, Allison LJ, Field KJ, Averill-Murray RC, Shaffer HB (2020) Individual heterozygosity predicts translocation success in threatened desert tortoises. Science 370:1086–1089
Simmons LW, Beveridge M, Wedell N, Tregenza T (2006) Postcopulatory inbreeding avoidance by female crickets only revealed by molecular markers. Mol Ecol 15:3817–3824
Slatyer RA, Mautz BS, Backwell PRY, Jennions MD (2012) Estimating genetic benefits of polyandry from experimental studies, a meta-analysis. Biol Rev 87:1–33. https://doi.org/10.1111/j.1469-185X.2011.00182.x
Sonsthagen SA, Wilson RE, Underwood JG (2017) Genetic implications of bottleneck effects of differing severities on genetic diversity in naturally recovering populations, an example from Hawaiian coot and Hawaiian gallinule. Ecol Evol 7:9925–9934
Spooner FEB, Pearson G, Freeman R (2018) Rapid warming is associated with population decline among terrestrial birds and mammals globally. Glob Change Biol 24:4521–4531
Teizeria JC, Huber DD (2021) The inflated significance of genetic diversity in conservation genetics. Proc Natl Acad Sci USA 118(10):e2015096118. https://doi.org/10.1073/pnas.2015096118
Thibault KM, Brown JH (2008) Impact of an extreme climatic event on community assembly. Proc Natl Acad Sci USA 105:3410–3415
Torres-Florez JP, Hucke-Gaete R, Rosenbaum H, Figuero CC (2014) High genetic diversity in a small population, the case of Chilean blue whales. Ecol Evol 4:1398–1412
Tregenza T, Wedell N (2002) Polyandrous females avoid the cost of inbreeding. Nature 415:71–73
Ujvari B, Madsen T, Kotenko T, Olsson M, Shine R, Wittzell H (2002) Low genetic diversity threatens imminent extinction for the Hungarian meadow viper (Vipera ursinii rakosiensis). Biol Conserv 105:127–130
Ujvari B, Brown G, Shine R, Madsen T (2016) Flood and famine; climate induced collapse of a tropical predator-prey community. Func Ecol 30:453–458
Vacquier VD (1998) Evolution of gamete recognition proteins. Science 281:1995–1998
Vuarin P, Bouchard A, Lesobre L, Levêque G, Chalah T, Jalme MS et al. (2019) Post-copulatory sexual selection allows females to alleviate the fitness costs incurred when mating with senescing males. Proc R Soc B 286:20191675
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7:167–184
Waples RS, Tiago Antao T, Luikart G (2014) Effects of overlapping generations on linkage disequilibrium estimates of effective population size. Genetics 197:769–780
White GC, Burnham KP (1999) Program MARK: survival estimation from populations of marked animals. Bird Study 46:S120–S139
Zhang B-Y, Hu H-Y, Song C-M, Huang K, Dunn DW, Yang X et al. (2020) MHC-based mate choice in wild golden snub-nosed monkeys. Front Genet 11:609414
Zimmerman SJ, Aldridge CL, Oyler-McCance SJ (2020) An empirical comparison of population genetic analyses using microsatellite and SNP data for a species of conservation concern. BMC Genomics 21:382
Acknowledgements
We are very grateful for comments on a previous version made by the associate editor and four reviewers which greatly improved our manuscript. The assistance provided by R.S. Waples that has been instrumental when preparing the manuscript and our analyses of Ne. We would also like to express our gratitude to B. Stille, H. Anderberg, L. Anderberg and C. Anderberg for their field work assistance. The study was financed by private funds.
Author information
Authors and Affiliations
Contributions
TM initiated and conceived the study. Field work was conducted by TM and BU. DB performed the population dynamics analyses. AG performed the molecular genetic analyses. BG performed the estimates of effective population size. MK performed the modelling analyses. TM wrote the manuscript with significant contributions from all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Ethical approval was received from the Skåne County Administrative Board in Dnr522-19974-2014, 178-201, 1287.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor Lounês Chikhi.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Madsen, T., Ujvari, B., Bauwens, D. et al. Polyandry and non-random fertilisation maintain long-term genetic diversity in an isolated island population of adders (Vipera berus). Heredity 130, 64–72 (2023). https://doi.org/10.1038/s41437-022-00578-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00578-2
This article is cited by
-
Is promiscuity the key? Multiple paternity in the garden dormouse (Eliomys quercinus)
Mammalian Biology (2024)
