
Abstract
Microbes that protect against infection inhabit hosts across the tree of life. It is unclear whether and how the host immune system may affect the formation of new protective symbioses. We investigated the transcriptomic response of Caenorhabditis elegans following novel interactions with a protective microbe (Enterococcus faecalis) able to defend against infection by pathogenic Staphylococcus aureus. We have previously shown that E. faecalis can directly limit pathogen growth within hosts. In this study, we show that colonisation by protective E. faecalis caused the differential expression of 1,557 genes in pathogen infected hosts, including the upregulation of immune genes such as lysozymes and C-type lectins. The most significantly upregulated host lysozyme gene, lys-7, impacted the competitive abilities of E. faecalis and S. aureus when knocked out. E. faecalis has an increased ability to resist lysozyme activity compared to S. aureus, suggesting that the protective microbe could gain a competitive advantage from this host response. Our finding that protective microbes can benefit from immune-mediated competition after introduction opens up new possibilities for biocontrol design and our understanding of symbiosis evolution. Crosstalk between the host immune response and microbe-mediated protection should favour the continued investment in host immunity and avoid the potentially risky evolution of host dependence.
Similar content being viewed by others

Introduction
Microbes that defend against infection by pathogens (along with parasites and parasitoids) colonise a large diversity of plant and animal hosts (Ford and King 2016; Kaltenpoth and Engl 2014; May and Nelson 2014; Parker et al. 2011). Protection can occur when symbiotic microbes suppress pathogen invasion by competing for resources/space or producing antimicrobial compounds (King 2019; Vorburger et al. 2013). Host-associated microbes can also suppress infection by modulating the host’s immune response to their benefit (Gerardo and Parker 2014). This immune-mediated competition can occur via the upregulation of host immune genes or components that play a role in disproportionately limiting the infection of pathogenic microbes over protective ones (Gerardo and Parker 2014; Mejía et al. 2014). This mechanism has been observed in animal-microbe symbioses. For example, the mosquito microbiome can modify basal immunity by upregulating immune genes conferring resistance to malaria parasites (Dong et al. 2009). Similarly, the commensal skin microbe, Staphylococcus epidermis, upregulates host immune responses that correlate with increased resistance to Staphylococcus aureus (Pastar et al. 2020). Immune-mediated competition has also been shown to structure diverse pathogen populations or communities by altering the competitive abilities of particular pathogen genotypes (Habets et al. 2012; Raberg et al. 2006; Ulrich and Schmid-Hempel 2012) or species (Bjørnstad and Harvill 2005; Lysenko et al. 2005; Margolis et al. 2010), respectively. The extent to which this mechanism of competition can operate at the origin of protective symbioses is unclear, particularly when protective microbes are invading a host species for the first time. Components of the host immune system—such as Toll- and NOD-like receptors, lysozymes and antimicrobial peptides—are known to regulate microbiota in Hydra (Bosch 2013), insects (Marra et al. 2021; Ryu et al. 2008), bobtail squid (Chen et al. 2017) and the mammalian gut (Mergaert 2018; Vaishnava et al. 2011), however there is a need for more information across diverse taxa and of the host control mechanisms specific to protective symbioses.
Interest in the mechanisms underpinning microbe-mediated protection has been surging because of its potential applicability in public health (O’Neill et al. 2018), species conservation (Trevelline et al. 2019) and agriculture (Singh et al. 2016). Although protective symbioses form naturally (Chrostek et al. 2017; Heath et al. 1999; Huigens et al. 2004; Jaenike et al. 2007), their artificial creation is being rapidly pursued for the biocontrol of infectious disease, either by introducing existing symbionts into new hosts (Bull and Turelli 2013) or by generating new symbionts via paratransgenesis (Magalhaes et al. 2019; Wang et al. 2017; Wilke and Marrelli 2015). For example, Aedes aegypti mosquitoes have been artificially infected with strains of the inherited symbiont Wolbachia that inhibit the transmission of arboviruses (Bian et al. 2013; Hoffmann et al. 2015; O’Neill et al. 2018). In the Wolbachia - mosquito system, the novel interaction upregulates host immune pathways and in turn, enhances antiviral protection (Rancès et al. 2012). It has been shown that such immune-upregulation can increase Wolbachia load (Pan et al. 2018), suggesting that immune-mediated competition could be occurring. Immune-mediated competition could facilitate the symbiont’s maintenance (Matthews et al. 2019) by allowing higher densities of symbiont to colonise. Higher densities of symbionts frequently correlate with greater protection (Drew and King 2022) and may feedback to promote maintenance of the symbiont. Over evolutionary time, the contribution of the host immune response may dictate the hosts vulnerability to pathogen infection should the protective symbiosis break down. Such break downs can be driven by imperfect transmission (Oliver et al. 2014), altered environment (Oliver et al. 2014; Raymann et al. 2017) or loss of protective traits (Alizon et al. 2013; Chrostek and Teixeira 2015; Ford and King 2016; Frank 1996; May and Nelson 2014). If protective microbes directly suppress infection, there is less need for hosts to defend themselves. This outcome could result in the divestment of costly host-based immune or damage response systems over evolutionary time (Ford and King 2016; Martinez et al. 2016; Metcalf and Koskella 2011; Parker et al. 2011). Conversely, host immune-mediated competition between protective and pathogenic microbes could maintain selection for host-based immunity (Mejía et al. 2014).
In this study, we investigated how a protective bacterial species (Enterococcus faecalis) could impact the transcriptomic response, and particularly the immune/defence responses, of novel Caenorhabditis elegans hosts to the pathogen, Staphylococcus aureus. These bacterivorous animals are genetically tractable with available immune knock-out mutants, and are thus useful for exploring the role of the immune response upon bacterial colonisation and infection. Enterococcus faecalis can be a pathobiont, ranging from being a gut commensal to opportunistic pathogen, and has been found to be protective in other animal hosts (Heikkilä and Saris 2003; Martín-Platero et al. 2006). It can colonise C. elegans guts through ingestion (Garsin et al. 2001). We have previously shown that E. faecalis can increase host survival during virulent infection by the opportunistic pathogen Staphylococcus aureus (King et al. 2016). Although we have demonstrated that this protective bacterium can quickly evolve to produce antimicrobial reactive oxygen species at levels to suppress S. aureus infection (King et al. 2016), other mechanisms are also at play. During co-colonisation, E. faecalis can inhibit S. aureus growth by stealing iron-binding siderophores that the pathogen releases into the environment (Ford et al. 2016). While neither E. faecalis nor S. aureus are known members of the native C. elegans microbiome, this tripartite system allows us to study how host and microbes interact at the inception of a novel symbiosis.
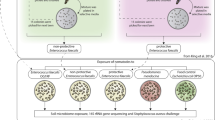
We exposed populations of C. elegans to bacteria for 12 h to compare host transcriptional responses to: (a) S. aureus infection in either the absence or presence of protective E. faecalis (Fig. 1a); (b) the ‘food’ bacterium Escherichia coli OP50 or E. faecalis alone (Fig. 1b); and (c) the ‘food’ bacterium E. coli OP50 or S. aureus alone (Fig. 1c). These bacterial colonisers are thus horizontally acquired. Using RNA-sequencing, we found that the presence of E. faecalis in S. aureus-infected C. elegans caused the differential expression of 1557 genes in infected hosts, including the upregulation of immune genes that encode for lysozymes and C-type lectins. E. faecalis is reported to be much more robust to attack by host lysozymes, with a minimum inhibitory concentration (MIC) of >62.5 mg/ml lysozyme (Varahan et al. 2013), whilst S. aureus has lower reported MICs of 15 mg/ml (Cisani et al. 1982). We therefore hypothesised that lysozyme expression may facilitate immune-mediated competition between pathogen and protector. By using a host with one of the most highly upregulated lysozyme genes (lys-7) knocked-out, we show that lysozyme expression grants a competitive advantage to E. faecalis in pathogen-infected hosts. Expression of lys-7 disproportionately suppresses the pathogen S.aureus, and as a result, facilitates enhanced protection by E. faecalis. This study shows that host immune responses can dictate the success of a protective microbe species when establishing in a novel competitive niche. Our work also highlights lysozymes as promising targets for manipulating microbe – microbe interactions within a host.

Young adult worms were exposed to bacteria for 12 h to compare host transcriptional response between: (a) S. aureus in the absence vs. presence of E. faecalis; (b) E. coli OP50 (food control) vs. E. faecalis; and (c) E. coli OP50 (food control) vs. S. aureus. We included five replicate populations for each unique treatment (i.e. only a single set of five replicates were completed for S. aureus and E. coli, despite being shown twice in the figure). After 12 h exposure, the RNA from ~1000 worms per sample was sequenced.
Materials and methods
Nematode host and bacteria
Caenorhabditis elegans is a nematode that ingests microbes for nutrients (Cabreiro and Gems 2013; Clark and Hodgkin 2014; Félix and Braendle 2010; Petersen et al. 2015) and is a well-established model for studying microbial colonisation and pathogenesis (Gravato-Nobre and Hodgkin 2005; Hope 1999; Montalvo-Katz et al. 2013; Peleg et al. 2008; Portal-Celhay and Blaser 2012). We used the simultaneous hermaphroditic N2 wild-type C. elegans strain from the Caenorhabditis Genetics Centre (CGC, University of Minnesota) along with a knock-out mutant for the lys-7 gene that is outcrossed into the N2 genetic background (strain CB6738, CGC). We generated genetically homogenous lines by selfing a single hermaphrodite for five generations and froze them in 50% M9 solution and 50% liquid freezing solution in cryotubes at −80 °C (Hope 1999). We regularly resurrected populations throughout experimentation to prevent the accumulation of de novo mutations in host populations. Worms were maintained at 20 °C on 9 cm nematode growth medium (NGM) plates seeded with Escherichia coli OP50. E. coli OP50 is grown at 30 °C shaking at 200 rpm overnight in lysogeny broth (LB) and 100 µl of this is spread onto NGM plates and incubated overnight at 30 °C (Hope 1999). To ensure clean stocks and to synchronise the life stages of populations for experimentation, we treated worms with bleach (NaClO) and sodium hydroxide (NaOH) solution which kills everything except unhatched eggs (Hope 1999).
We used Staphylococcus aureus strain MSSA476 (GenBank: BX571857.1), an invasive community-acquired methicillin-susceptible isolate, as the pathogen in our system. C. elegans likely encounter Staphylococcus species in their natural habitat (Montalvo-Katz et al. 2013; Rossouw and Korsten 2017), however specific interactions with S. aureus here are considered novel as this species has not been recorded interacting with natural populations of C. elegans. As the protective microbe, we used Enterococcus faecalis OG1RF (GenBank: CP002621.1), an isolate from the human digestive tract. E. faecalis is naturally protective in a variety of animals (Heikkilä and Saris 2003; Martín-Platero et al. 2006) but is not a known member of the C. elegans native microbiome. Both E. faecalis and S. aureus are horizontally acquired by C. elegans through ingestion (Garsin et al. 2001; Sifri et al. 2003). We grew each species from a single colony overnight in 6 ml Todd Hewitt Broth (THB) shaking at 200 rpm at 30 °C. Bacteria were frozen in a 1:1 ratio of sample to 50% glycerol solution in cryotubes at −80 °C.
Experimental set-up and RNA extraction
We exposed populations of young adult worms to bacteria for 12 h to compare host transcriptional responses to: (a) S. aureus in the absence vs. presence of E. faecalis (Fig. 1a); (b) E. coli OP50 (food control) vs. E. faecalis (Fig. 1b); and (c) food control vs. S. aureus (Fig. 1c). Five replicate populations were used for each unique treatment. We collected sterile and age-synchronised eggs using the bleach-sodium hydroxide solution. We kept these eggs in M9 buffer without food, shaking for ~8 h at 88 rpm and 20 °C to arrest development at L1. We transferred ~5000 L1 worms per replicate population to 9 cm NGM plates seeded with E. coli and placed at 20 °C to grow for 2 days. We chose this density to avoid overcrowding and starvation. Bacteria were grown from frozen culture overnight in 6 ml culture (THB for E. faecalis and S. aureus, LB for E. coli OP50) in a shaking incubator at 30 °C at 200 rpm and standardised their optical density to OD600 of 1 which corresponds to ~1 × 109 cells/mL. We spread 120 μl culture per species onto 9 cm Tryptic Soy Broth (TSB) agar plates and incubated them overnight at 30 °C. Where worms were to be exposed to both E. faecalis and S. aureus, we mixed 120 μl of each culture on the same TSB plate. We removed young adult stage worms from the NGM plates, washed them in 50 ml M9 buffer five times and placed ~2000 young adults on the exposure plates at 25 °C for 12 h. At no time did the worms ever run out of bacteria to eat.
After 12 h of exposure, we washed worms off each plate using M9 buffer within 10 min in an order determined by a random number generator. We chose 12 h of exposure to avoid host mortality but provide sufficient time for C. elegans to respond to bacterial exposure and infection. We washed the worms in 10 ml M9 buffer five times and put ~1000 worms in 50 μl into Eppendorf tubes containing 1 ml Trizol and vortexed for 20 s. We then freeze-thawed the samples of worms three times using dry ice and a heat block (40 °C) to break the worm cuticle and stored at −80 °C. We extracted RNA using Zymo spin columns, following the manufacturer’s instructions with on-column DNA digestion using DNase I.
RNA sequencing
We checked the quality of the RNA using an Agilent 2100 Bioanalyzer with the Eukaryote total RNA pico chip. We quantified the resulting RNA using the Qubit® Fluorometer (Invitrogen) and all samples were diluted to the same final concentration. This was done to ensure that each sample is represented evenly for accurate transcript quantification. The Oxford Genomics Centre then performed library preparation and sequencing. The polyA signal was used to select the mRNA fraction from the RNA to capture just C. elegans and not the bacteria from the sample. RNA was then converted to cDNA. Second strand cDNA synthesis incorporated dUTP and the cDNA was then end-repaired, A-tailed and adapter-ligated. Prior to amplification, samples underwent uridine digestion. The prepared libraries were size selected, multiplexed and checked for quality before paired-end sequencing using NovaSeq6000 with 150 bp paired-end reads. On average, each replicate sample generated 22,588,105 reads, of which 82.55% were assigned to C. elegans and 4.46% to bacteria.
SortMeRNA (Kopylova et al. 2012) was used for further filtering of ribosomal RNA prior to alignment, on average each sample had 1.26% reads mapping to small, and 4.65% to large, subunit eukaryote rRNA sequences. Full detail on the number of reads associated with each run are shown in SI File 5.
We checked raw reads for quality using FastQC (0.11.5). Current (release 96) GTF and cDNA FASTA files were downloaded from the ensemble database for C. elegans (WBcel235 version of the C. elegans reference genome). We created a transcript index using Kallisto and the C. elegans WBcel235 cDNA FASTA file. We then performed pseudoalignment using Kallisto with 100 bootstraps and calculated transcript abundances. Kallisto pseudoalignment is a fast and accurate way of quantifying transcript abundance since it matches sequences to existing transcripts from a reference library, rather than performing a full alignment to the genome. This means that it tells you ‘what’ transcript a sequence is compatible with, but not ‘where’. These data are sufficient for transcript abundance quantification. We then used the R package Sleuth to perform statistical analyses (see ‘statistical analysis’).
Host mortality
We tested whether the lys-7 gene played a role in host mortality in response to S. aureus and E. faecalis colonisation and co-colonisation. We grew age-synchronised eggs to young adult stage on 9 cm NGM plates seeded with a lawn of E. coli OP50. Using the same protocols as describe above, we grew E. coli OP50, S. aureus and E. faecalis in vitro overnight, standardised the cultures to OD600 of 1, and made the exposure plates. We washed ~250 young adults of either CB6738 (lys-7 knock-out) or N2 wild-type in 50 ml M9 five times before exposing them at 25 °C for 24 h. The proportion of dead worms were then counted as a measure of host mortality.
Bacterial colonisation
We measured the co-colonisation ability of E. faecalis and S. aureus in wild-type worms and lys-7 gene knockout worms. We exposed CB6738 (lys-7 knock-out) and N2 wild-type worms to both S. aureus and E. faecalis together following the same protocol as above (see ‘Host mortality’). After 24 h of exposure to the bacteria, we collected 7–10 live worms per exposure plate and washed them in 5 ml of M9 five times under the microscope. To release the colonising bacteria, we placed the worms in 2 ml screwcap tubes with 50 μl M9 and 1.5 mm Zirconium beads (Benchmark Scientific) and broke the cuticle by shaking the tubes at 320 rpm for 45 s. We plated serial dilutions onto Mannitol Salt Agar to isolate S. aureus and TSB with 100 μg/ml rifampicin (Sigma-Aldrich) to isolate E. faecalis. We incubated the plates at 30 °C overnight before counting the number of colony-forming units (CFUs) per host.
Statistical analysis
We performed differential expression analysis on the transcript abundance outputs from Kallisto using Sleuth in R v 3.2.0 (http://www.r-project.org/) and following the same format as previously published work on this system (Ford and King 2021). Sleuth uses generalised linear models to identify coefficients of the strength of expression. To identify differentially expressed transcripts between two experimental treatments, Sleuth compares the full model to a reduced model that assumes abundances are the same across the treatments (Pimentel et al. 2017), we then ran likelihood ratio tests of fitted models. This process was completed for three comparisons (S. aureus in the absence vs. presence of E. faecalis, OP50 vs. E. faecalis and OP50 vs. S. aureus) with treatment modelled as the dependent variable. SI files 1, 3 and 4 show DEG results from sleuth models and LRTs. The significance of treatment was determined by a q value of <0.05 (p value adjusted by means of the Benjamini-Hochberg false discovery date, FDR, correction for multiple comparisons). We performed a gene ontology (GO) term enrichment analysis on the significant differentially expressed genes using the g:Profiler online tool with the Benjamini-Hochberg FDR correction for multiple comparisons (Raudvere et al. 2019).
We used parametric tests for all data which met the required assumptions. We used the Shapiro test to detect whether data was normally distributed and F-tests to compare the variances of two samples from normal populations. We compared bacterial CFUs per host using two-sample t-tests. We used a binomial GLM to compare host mortality among host strains colonised by E. faecalis and E. coli OP50. Models were checked for overdispersion via the ratio of residual deviance to residual degrees of freedom. To account for overdispersion, we used quasibinomial GLMs to compare mortality among co-colonised host strains and host strains colonised by S. aureus singularly. In all mortality models the proportion of dead worms was modelled as a function of host strain (lys-7 mutant or wild-type N2). We assessed the fit of GLMs by checking deviance values and diagnostic plots of residuals and quantile-quantiles. Hosmer-Lemershow goodness of fit tests were used to check there were no significant differences between the fitted model and the observed data. We corrected p-values where multiple comparisons were made from the same dataset using the FDR (Benjamini and Hochberg) method.
Results
Gut colonisation by E. faecalis alters host response to virulent infection
We used RNA-sequencing to assess the transcriptional changes in C. elegans hosts during infection by virulent S. aureus, in the presence (co-colonisation) vs. absence (single colonisation) of E. faecalis (Fig. 1a). We found that the presence of E. faecalis drove significant differential expression of 1557 nematode genes compared with E. faecalis’ absence (Fig. 2, Supplementary File 1). Of these genes, 521 were downregulated, whilst 1036 were upregulated. We performed a GO-term enrichment analysis on this list of genes and identified significantly enriched GO-terms in biological processes including “defence response to bacterium” (GO:0042742, FDR-adjusted P = 6.12E−09, Supplementary File 2) and “innate immune response” (GO:0045087, FDR-adjusted P = 4.09E−12, Supplementary File 2). We also found significantly enriched GO-terms in molecular functions including ‘structural constituent of cuticle’ (FDR-adjusted P = 1.02E−37, Supplementary File 2).

a Differentially expressed genes (DEGs) of host C. elegans under single colonisation (orange) by E. faecalis and S. aureus, relative to co-colonisation (green) whereby E. faecalis protects against the S. aureus pathogen. The 46 immune gene families differentially regulated by E. faecalis-mediated protection are detailed in the green box, blue arrows indicate downregulation and red upregulation. See supplementary files 1, 3, 4 and 7 for further detail on DEGs across treatments. b Gene ontology (GO) terms significantly enriched in the list of C. elegans genes differentially regulated under co-colonisation and Enterococcus faecalis-mediated protection. Enrichment analysis performed using g:Profiler with Benjamini-Hochberg FDR correction for multiple comparisons.
We found that the enriched GO-terms (Fig. 2b). relating to defence and the immune response were linked to numerous differentially expressed lysozyme-encoding genes (Fig. 2a). Lysozymes are important in the host defence response to bacteria as these enzymes break down bacterial cell walls (Ragland and Criss 2017). We found that the presence of E. faecalis in infected hosts was associated with the upregulation of the invertebrate lysozyme genes, ilys-2 and ilys-5 compared to when hosts were only infected by S. aureus (ilys-2 beta = 1.171, P = 6.28E−06; ilys-5 beta = 0.85, P = 0.00055). We also found a significant upregulation in the lysozyme genes, lys-1 (beta = 0.77, P = 0.00028), lys-2 (beta = 0.78, P = 3.01E−05), lys-7 (beta = 2.12, P = 1.15E−07) and lys-10 (beta = 3.46, P = 4.36E−05). Only lys-3 was significantly downregulated (beta = −1.2, P = 0.00025). The most significantly upregulated lysozyme gene was lys-7. By measuring how E. faecalis alone alters host transcription compared to an E. coli food control (Fig. 1b, Supplementary File 3), we found that of these lysozyme genes, E. faecalis only upregulated lys-1 (beta = 0.017, P = 0.0395). This result suggests that E. faecalis colonisation alone is sufficient to upregulate lys-1 in nematodes, whilst lys-2, lys-7 and lys-10 also require pathogen presence to be upregulated. By measuring how S. aureus infection alone alters host transcription compared to an E. coli food control (Fig. 1c, Supplementary File 4), we found that of these lysozyme genes, S. aureus only caused the downregulation of lys-1 (Y22F5A.4.1, beta = −1.0, P = 0.000009; Y22F5A.4.2, beta = 0.033, P = −0.92) and left the others unaffected. This result demonstrates that the presence of E. faecalis and S. aureus appears to impact lys-1 in opposing directions whilst lys-2, lys-7 and lys-10 in nematodes require the co-colonisation of E. faecalis and S. aureus for upregulation.
In addition to lysozymes, the enriched GO-terms relating to defence and the immune response were connected with 28 differentially expressed C-type lectin (clec) transcripts (Fig. 2). Colonisation of E. faecalis during pathogen infection was associated with the upregulation of 20 clec genes and the downregulation of a further seven clec genes compared to when hosts were infected by only S. aureus (Fig. 1a, Fig. 2). These genes are carbohydrate-binding proteins that play a host defensive role against gram-positive bacteria (Pees et al. 2017). By comparing the impact of E. faecalis alone on host gene expression to gene expression in E. coli control hosts (Fig. 1b, Supplementary File 3), we found that colonisation by this bacterium was not sufficient to explain these results. Some of these nematode clec genes were not differentially expressed by E. faecalis alone, whilst clec-41 (downregulated) and clec-146 (Y48E1B.9b.1, upregulated) were regulated in the opposite direction to that seen during pathogen infection. It therefore appears that the expression profile of the 28 clec transcripts cannot be caused by E. faecalis colonisation alone, but is likely shaped by E. faecalis and S. aureus co-colonisation (Supplementary Files 3 and 4).
The enriched GO-terms relating to structural constituents of the cuticle were linked to many differentially expressed cuticular collagen genes (Supplementary File 2). We found that colonisation by both E. faecalis and S. aureus was associated with the upregulation of 44 col and 8 dpy genes compared to when hosts were only infected by S. aureus (Fig. 1a, Supplementary File 1). Both col and dpy genes are upregulated in response to pathogens which produce extracellular proteins that degrade collagen and weaken the host hypodermis (Sellegounder et al. 2019). This process facilitates pathogen invasion of the host and can be important for resistance to multiple pathogens, including S. aureus MSSA476 (Sellegounder et al. 2019). By comparing the transcriptional response of C. elegans in response to colonisation by E. faecalis alone versus a food control (Fig. 1b, Supplementary File 3), we discovered that E. faecalis appears to be sufficient to upregulate some col genes (col-17, col-88, col-97 and col-167, Supplementary File 3). Of these, we found that S. aureus alone does not cause the upregulation of col-17 or col-88, but does upregulate col-97 and col-167 compared to when nematodes are exposed to a food control (Supplementary File 4). This result shows that two upregulated cuticular collagen genes (col-17 and col-88) are upregulated solely by E. faecalis colonisation, whilst another two can be upregulated by either bacterium (col-97 and col-167). Upregulation of the remaining genes is likely to require the coinfection of E. faecalis and S. aureus. However, we do not exclude the possibility that the host transcriptomic response observed under coinfection may be affected by an overall higher dosage of bacteria (combined dose of E. faecalis and S. aureus).
Lysozyme expression determines outcome of competition between protective microbes and pathogens
Lysozymes are a well-known conserved antimicrobial defence (Ragland and Criss 2017). Because E. faecalis is more resistant to lysozyme activity than S. aureus (Cisani et al. 1982; Varahan et al. 2013), we hypothesised that lysozyme expression would disproportionately impact their competitive ability. Lysozymes could thereby contribute to host immune-mediated competition between the bacterial species and give protective E. faecalis an advantage. We focused on the impact of the most significantly differentially expressed lysozyme gene, lys-7, on bacterial colonisation and protection.
We first tested whether knocking out lys-7 increased host mortality to E. faecalis, S. aureus and a E.coli OP50 (food-only) control. We expected lys-7 knock-out hosts to have increased mortality compared to wild-type N2 hosts and that this mortality would be much higher with the pathogen, S. aureus, than E. faecalis. As expected, knocking out lys-7 increased host mortality across all bacterial exposures, however this increase was only small in the E. faecalis treatment and E.coli food control as these bacteria have very low virulence in this system (Fig. 3. E. coli control: Binomial GLM, df = 1, FDR-corrected P = 0.04; E. faecalis: Binomial GLM, df = 1, FDR-corrected P = 0.04; S. aureus: Quasibinomial GLM, F = 11.2, df = 1, FDR-corrected P = 0.03). In the lys-7 knock-out, host mortality after 24 h exposure to food or E. faecalis remained very small (from 0% in N2 to 0.4%, and from 0.2% in N2 to 1.2%, respectively), but mortality caused by S. aureus infection increased greatly (from 57.6% in N2 to 90.3% in lys-7 knockout nematodes). We then tested whether knocking out lys-7 increased host mortality when co-colonised by S. aureus and E. faecalis. We found that host mortality was higher in lys-7 knock-out hosts than in the wild-type (Fig. 3d. Quasibinomial GLM. host strain: F = 14.7, df = 1, P = 0.05). The protective effect of E. faecalis was maintained in lys-7 knock-outs, with mortality dropping from 90.3% under S.aureus colonisation to 24.9% under co-colonisation with E.faecalis (Quasibinomial GLM. Bacterial colonisation: F = 192.9, df = 1, P < 0.01).

Percentage mortality of the lys-7 knock-out mutant and wild-type N2 C.elegans host after single colonisation (orange) by food control E. coli (a), protective E. faecalis (b) and pathogenic S. aureus (c) and after co-colonisation (green) whereby E. faecalis protects against pathogenic S. aureus (d). Within-host bacterial density in colony-forming units (CFUs) of E. faecalis (e), and S. aureus (f) after 24 h of co-colonisation. Each treatment was replicated five independent times and 7–10 worms were collected per replicate for CFU quantification. *P < 0.05, **P < 0.01, ***P < 0.001.
To investigate whether lys-7 expression disproportionately impacts S. aureus colonisation over E. faecalis colonisation, we measured their CFUs during co-colonisation. We found a trend towards lower S. aureus infection loads in the wild-type host compared to in the lys-7 knock-out mutant (Fig. 3f. t-test, t = 1.42, df = 8, P = 0.19). Although not significant, the results are consistent with our mortality data. By contrast, we found that E. faecalis was significantly better at colonising the wild-type host than the lys-7 knock-out mutant (Fig. 3e. t-test, t = −3.5, df = 8, P = 0.0076).
Discussion
Protective host-microbe symbioses can form naturally (Chrostek et al. 2017; Heath et al. 1999; Huigens et al. 2004; Jaenike et al. 2007), but are also artificially forged in an effort to tackle the spread of infectious disease (Bull and Turelli 2013; Magalhaes et al. 2019; Wang et al. 2017; Wilke and Marrelli 2015). In an emergent symbiosis, the host immune system may have a key role in allowing protective microbes to invade and be maintained. We investigated the effect of a novel protective microbe on the transcriptomic response of Caenorhabditis elegans hosts to pathogen infection. Neither the pathogen species (S. aureus), nor the protective microbe (E. faecalis), have been recorded in the native C. elegans microbiome, making the system valuable for studying the inception of a tripartite interaction. Under co-colonisation of protective microbe and pathogen we observed the upregulation of many nematode immune genes such as those coding for the production of antimicrobial lysozymes and C-type lectins (Dierking et al. 2016). This result reflects immune-related gene expression patterns in hosts that are naturally colonised by protective microbes (Mejía et al. 2014; Montalvo-Katz et al. 2013) as well as artificially established host-protective microbe systems (Rancès et al. 2012). The artificial symbiosis between Wolbachia and mosquitoes generates the upregulation of a suite of immune genes, including C-type lectins and defensin, that suppress Dengue and Chikungunya viruses (Moreira et al. 2009; Rancès et al. 2012). We also saw the upregulation of cuticular collagen genes, including col and dpy genes. Pathogens frequently produce extracellular proteins to degrade collagen in the host hypodermis (Koziel and Potempa 2013) and C. elegans appears to alter its cuticle structure in response (Coolon et al. 2009; Sellegounder et al. 2019; Wong et al. 2007). Upregulation of these collagen genes is essential for nematode defence against Pseudomonas aeruginosa, Salmonella enterica and S. aureus (Sellegounder et al. 2019), and has been implicated in responses to E. faecalis, Serratia marcescens and Photorhabdus luminescens (Wong et al. 2007).
Our results indicate that the nematode immune system might alter the within-host competition of E. faecalis and S. aureus, benefitting the former’s colonisation. Lysozymes are key elements of the innate immune system and catalyse the degradation of bacterial cell wall peptidoglycan and modulate the host immune response through the release of pattern recognition receptors (Ragland and Criss 2017). E. faecalis has been shown to be more resistant to lysozyme activity than S. aureus (Cisani et al. 1982), whilst lysozymes have also been found to contribute to nematode resistance against S. aureus (Kong et al. 2014; Visvikis et al. 2014). By knocking out lys-7, the most significantly upregulated lysozyme gene during co-colonisation, we found these worms had a significantly lower within-host load of protective E. faecalis. By contrast, there was a trend, albeit insignificant, for knock-out worms to have higher S. aureus colonisation. It is unclear if lys-7 could directly benefit E. faecalis growth. However, a small reduction in S. aureus colonisation could be sufficient to indirectly impact E. faecalis fitness through immune-mediated competition. It was previously demonstrated that S. aureus drives the downregulation of lys-7 expression in C. elegans and when expression is restored through chemical treatment, pathogen-induced host mortality decreased along with infection load (Kong et al. 2014). These results suggest that the upregulation of lysozyme genes in co-colonised hosts is likely to benefit E. faecalis’ ability to colonise, and in turn, confer other mechanisms of protection, such as ROS production (King et al. 2016) and siderophore exploitation (Ford et al. 2016). Increased colonisation by E. faecalis has previously been shown to relate to enhanced protection in our system (King et al. 2016; Rafaluk-Mohr et al. 2018) and in other protective symbioses more generally (Drew and King 2022).
We found that the presence of E. faecalis was sufficient to cause the upregulation of one lysozyme and two collagen genes. Colonisation by E. faecalis alone caused the upregulation of the lysozyme gene, lys-1, and the cuticular collagen genes, col-17 and col-88. Unlike this, we found that S. aureus alone drives the downregulation of the lys-1 gene. Thus, S. aureus and E. faecalis appear to have opposing effects on lysozyme expression, with E. faecalis dominating control over lys-1 during co-colonisation. The beneficial effect that lysozyme expression has for both E. faecalis and the host C. elegans suggests that E. faecalis-induced lysozyme expression could be maintained by selection in a pathogen rich environment. This mechanism of immune-mediated competition is similar to that between the commensal skin microbe, Staphylococcus epidermis, and harmful S. aureus (Pastar et al. 2020). The commensal upregulates host expression of an antimicrobial protein effective at killing the pathogen (Pastar et al. 2020), whilst S. aureus conversely downregulates the expression of the antimicrobial protein.
Immune upregulation caused by protective microbes might be costly to the host and thereby affect ability to spread within a host population (Oliver et al. 2008; Vorburger et al. 2013). Wolbachia strains introduced into novel mosquito hosts were found to upregulate host immunity, but not when in native mosquitoes. Although this upregulation enhanced protection (Rancès et al. 2012) and potentially Wolbachia density (Pan et al. 2018), the absence of this response in the native host suggests that its cost may outweigh its benefit (Rancès et al. 2012). When protection is inducible, rather than constitutive, it may reduce the cost to the host of maintaining the symbiont, making the symbiosis more likely to persist. In our study, we found many immune genes upregulated by E. faecalis in infected hosts only. These plastic changes in immune gene expression, or plastic divestment of immunity in a protective symbiosis (Martinez et al. 2016), might also reduce the cost of harbouring E. faecalis and prevent vulnerability to infection in future generations should the relationship break down (Ford and King 2021).
Despite the launching of host immune factors, our results suggest direct pathogen suppression from E. faecalis is likely still the primary means of host protection (Ford et al. 2016; King et al. 2016). Direct symbiont-pathogen interactions are common among natural protective symbioses (Ford and King 2016). Whilst lys-7 knockout worms were highly susceptible to S. aureus infection experiencing 90% mortality, worms suffered less mortality during co-colonisation. This result was consistent for both knockout and wild-type worms. Thus, without lys-7, E. faecalis still confers a degree of protection, in agreement with previous findings that E. faecalis competes directly with S. aureus for iron resources via iron-binding siderophores (Ford et al. 2016) and suppresses the pathogen via superoxides (King et al. 2016). While lys-7 was the focus of direct testing here, it is possible that other candidate genes could be contributing to protection. Further work should test other lysozyme-encoding genes (specifically lys-2 and lys-10), C-type lectin and collagen-encoding genes for a strengthened test of immune-mediated competition.
Immune-mediated competition ultimately selects for the persistence of those species best able to evade the host immune factors elicited (Bjørnstad and Harvill 2005; Habets et al. 2012; Lysenko et al. 2005; Margolis et al. 2010; Raberg et al. 2006; Ulrich and Schmid-Hempel 2012). This phenomenon has been predominately examined in host-parasite systems (Raberg et al. 2006), but may be relevant for symbioses more generally. Our study shows that this process may benefit the establishment of protective microbes upon introduction to novel hosts. Specifically, we find the protective microbe was given a competitive edge from lysozyme production in a novel host species. This result may have emerged because E. faecalis is widely distributed among animal microbiota—as a protector (Heikkilä and Saris 2003), commensal and pathogen (Mason et al. 2011)—and lysozymes are common components of innate immune systems. Bacterial adaptations to these immune components in one host species might therefore be relevant in another. Indeed, the transfer of protective microbes to new hosts is more successful between host species that are more phylogenetically similar, i.e. their immune systems may be more similar (Russell and Moran 2005). That said, immune-mediated competition in our study could have been a convenient by-product of a triggered immune response to a novel bacterium. Our results nevertheless highlight the complexity of interactions between hosts and their microbial communities. Artificially established symbioses between animals and microbes are set to become an important tool in biocontrol (Ford and King 2016).
Identifying host factors that govern the colonisation of protective microbes will provide vital targets for approaches such as RNA interference or drug development. These developments may allow us to encourage novel symbioses to form, or indeed to disrupt existing symbioses.
Data availability
Raw and processed transcriptomic data for C. elegans is publicly available in the Gene Expression Omnibus (GSE151731). The experimental data associated with the study is available at 10.6084/m9.figshare.c.6250053.
References
Alizon S, de Roode JC, Michalakis Y (2013) Multiple infections and the evolution of virulence. Ecol Lett 16(4):556–67
Bian G, Zhou G, Lu P, Xi Z (2013) Replacing a native Wolbachia with a novel strain results in an increase in endosymbiont load and resistance to dengue virus in a mosquito vector. PLoS Negl Trop Dis 7(6):e2250
Bjørnstad ON, Harvill ET (2005) Evolution and emergence of Bordetella in humans. Trends Microbiol 13(8):355–9
Bosch TC (2013) Cnidarian-microbe interactions and the origin of innate immunity in metazoans. Annu Rev Microbiol 67:499–518
Bull JJ, Turelli M (2013) Wolbachia versus dengue: Evolutionary forecasts. Evol Med Public Health 2013(1):197–207
Cabreiro F, Gems D (2013) Worms need microbes too: microbiota, health and aging in Caenorhabditis elegans. EMBO Mol Med 5(9):1300–10
Chen F, Krasity BC, Peyer SM, Koehler S, Ruby EG, Zhang X et al. (2017) Bactericidal permeability-increasing proteins shape host-microbe interactions. mBio 8:e00040–17
Chrostek E, Pelz-Stelinski K, Hurst GDD, Hughes GL (2017) Horizontal Transmission of Intracellular Insect Symbionts via Plants. Front Microbiol 8:2237
Chrostek E, Teixeira L (2015) Mutualism breakdown by amplification of Wolbachia genes. PLoS Biol 13(2):e1002065
Cisani G, Varaldo PE, Grazi G, Soro O (1982) High-level potentiation of lysostaphin anti-staphylococcal activity by lysozyme. Antimicrob Agents Chemother 21(4):531–5
Clark LC, Hodgkin J (2014) Commensals, probiotics and pathogens in the Caenorhabditis elegans model. Cell Microbiol 16(1):27–38
Coolon JD, Jones KL, Todd TC, Carr BC, Herman MA (2009) Caenorhabditis elegans genomic response to soil bacteria predicts environment-specific genetic effects on life history traits. PLOS Genet 5:e1000503
Dierking K, Yang W, Schulenburg H (2016) Antimicrobial effectors in the nematode Caenorhabditis elegans: an outgroup to the Arthropoda. Philos Trans R Soc Lond B Biol Sci 371:1695
Dong Y, Manfredini F, Dimopoulos G (2009) Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog 5(5):e1000423
Drew GC, King KC (2022) More or less? The effect of symbiont density in protective mutualisms. Am Nat 199(4):443–54
Ford SA, Kao D, Williams D, King KC (2016) Microbe-mediated host defence drives the evolution of reduced pathogen virulence. Nat Commun 7:13430
Ford SA, King KC (2016) Harnessing the Power of Defensive Microbes: Evolutionary Implications in Nature and Disease Control. PLoS Pathog 12(4):e1005465
Ford SA, King KC (2021) In Vivo Microbial Coevolution Favors Host Protection and Plastic Downregulation of Immunity. Mol Biol Evol 38(4):1330–1338
Frank SA (1996) Models of parasite virulence. Q Rev Biol 71(1):37–78
Félix MA, Braendle C (2010) The natural history of Caenorhabditis elegans. Curr Biol 20(22):R965–9
Garsin DA, Sifri CD, Mylonakis E, Qin X, Singh KV, Murray BE et al. (2001) A simple model host for identifying Gram-positive virulence factors. Proc Natl Acad Sci USA 98(19):10892–7
Gerardo NM, Parker BJ (2014) Mechanisms of symbiont-conferred protection against natural enemies: an ecological and evolutionary framework. Curr Opin Insect Sci 4:8–14
Gravato-Nobre MJ, Hodgkin J (2005) Caenorhabditis elegans as a model for innate immunity to pathogens. Cell Microbiol 7(6):741–51
Habets MG, Rozen DE, Brockhurst MA (2012) Variation in Streptococcus pneumoniae susceptibility to human antimicrobial peptides may mediate intraspecific competition. Proc Biol Sci 279(1743):3803–11
Heath BD, Butcher RD, Whitfield WG, Hubbard SF (1999) Horizontal transfer of Wolbachia between phylogenetically distant insect species by a naturally occurring mechanism. Curr Biol 9(6):313–6
Heikkilä MP, Saris PE (2003) Inhibition of Staphylococcus aureus by the commensal bacteria of human milk. J Appl Microbiol 95(3):471–8
Hoffmann AA, Ross PA, Rašić G (2015) Wolbachia strains for disease control: ecological and evolutionary considerations. Evol Appl 8(8):751–68
Hope IA (1999) C. elegans: a practical approach. Oxford University Press, Oxford
Huigens ME, de Almeida RP, Boons PA, Luck RF, Stouthamer R (2004) Natural interspecific and intraspecific horizontal transfer of parthenogenesis-inducing Wolbachia in Trichogramma wasps. Proc Biol Sci 271(1538):509–15
Jaenike J, Polak M, Fiskin A, Helou M, Minhas M (2007) Interspecific transmission of endosymbiotic Spiroplasma by mites. Biol Lett 3(1):23–5
Kaltenpoth M, Engl T (2014) Defensive microbial symbionts in Hymenoptera. Funct Ecol 28(2):315–27
King KC (2019) Quick guide: defensive symbionts. Curr Biol 29:R78–R80
King KC, Brockhurst MA, Vasieva O, Paterson S, Betts A, Ford SA et al. (2016) Rapid evolution of microbe-mediated protection against pathogens in a worm host. ISME J 10(8):1915–24
Kong C, Tan MW, Nathan S (2014) Orthosiphon stamineus protects Caenorhabditis elegans against Staphylococcus aureus infection through immunomodulation. Biol Open 3(7):644–55
Kopylova E, Noé L, Touzet H (2012) SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 14(24):3211–17
Koziel J, Potempa J (2013) Protease-armed bacteria in the skin. Cell Tissue Res 351:325–37
Lysenko ES, Ratner AJ, Nelson AL, Weiser JN (2005) The role of innate immune responses in the outcome of interspecies competition for colonization of mucosal surfaces. PLoS Pathog 1(1):e1
Magalhaes T, Bergren NA, Bennett SL, Borland EM, Hartman DA, Lymperopoulos K et al. (2019) Induction of RNA interference to block Zika virus replication and transmission in the mosquito Aedes aegypti. Insect Biochem Mol Biol 111:103169
Margolis E, Yates A, Levin BR (2010) The ecology of nasal colonization of Streptococcus pneumoniae, Haemophilus influenzae and Staphylococcus aureus: the role of competition and interactions with host’s immune response. BMC Microbiol 10:59
Marra A, Hanson MA, Kondo S, Erkosar B, Lemaitre B (2021) Drosophila Antimicrobial Peptides and Lysozymes Regulate Gut Microbiota Composition and Abundance. mBio 12(4):e0082421
Martinez J, Cogni R, Cao C, Smith S, Illingworth CJ, Jiggins FM (2016) Addicted? Reduced host resistance in populations with defensive symbionts. Proc Biol Sci 283:1833
Martín-Platero AM, Valdivia E, Ruíz-Rodríguez M, Soler JJ, Martín-Vivaldi M, Maqueda M et al. (2006) Characterization of antimicrobial substances produced by Enterococcus faecalis MRR 10-3, isolated from the uropygial gland of the hoopoe (Upupa epops). Appl Environ Microbiol 72(6):4245–9
Mason KL, Stepien TA, Blum JE, Holt JF, Labbe NH, Rush JS et al. (2011) From commensal to pathogen: translocation of Enterococcus faecalis from the midgut to the hemocoel of Manduca sexta. MBio 2(3):e00065–11
Matthews AC, Mikonranta L, Raymond B (2019) Shifts along the parasite-mutualist continuum are opposed by fundamental trade-offs. Proc Biol Sci 286(1900):20190236
May G, Nelson P (2014) Defensive mutualisms: do microbial interactions within hosts drive the evolution of defensive traits? Funct Ecol 28(2):356–63
Mejía LC, Herre EA, Sparks JP, Winter K, García MN, Van Bael SA et al. (2014) Pervasive effects of a dominant foliar endophytic fungus on host genetic and phenotypic expression in a tropical tree. Front Microbiol 5:479
Mergaert P (2018) Role of antimicrobial peptides in controlling symbiotic bacterial populations. Nat prod Rep. 35(4):336–56
Metcalf CJE, Koskella B (2019) Protective microbiomes can limit the evolution of host pathogen defense. Evol Lett 3:534–43
Montalvo-Katz S, Huang H, Appel MD, Berg M, Shapira M (2013) Association with soil bacteria enhances p38-dependent infection resistance in Caenorhabditis elegans. Infect Immun 81(2):514–20
Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu G, Pyke AT, Hedges LM et al. (2009) A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell 139(7):1268–78
O’Neill SL, Ryan PA, Turley AP, Wilson G, Retzki K, Iturbe-Ormaetxe I et al. (2018) Scaled deployment of Wolbachia to protect the community from Aedes transmitted arboviruses. Gates Open Res 2:36
Oliver KM, Campos J, Moran NA, Hunter MS (2008) Population dynamics of defensive symbionts in aphids. Proc Biol Sci 275(1632):293–9
Oliver KM, Smith AH, Russell JA (2014) Defensive symbiosis in the real world \‘96 advancing ecological studies of heritable, protective bacteria in aphids and beyond. Funct Ecol 28(2):341–55
Pan X, Pike A, Joshi D, Bian G, McFadden MJ, Lu P et al. (2018) The bacterium Wolbachia exploits host innate immunity to establish a symbiotic relationship with the dengue vector mosquito Aedes aegypti. ISME J 12(1):277–88
Parker BJ, Barribeau SM, Laughton AM, de Roode JC, Gerardo NM (2011) Non-immunological defense in an evolutionary framework. Trends Ecol Evol 26(5):242–8
Pastar I, O’Neill K, Padula L, Head CR, Burgess JL, Chen V et al. (2020) Staphylococcus epidermidis Boosts Innate Immune Response by Activation of Gamma Delta T Cells and Induction of Perforin-2 in Human Skin. Front Immunol 11:550946
Pees B, Kloock A, Nakad R, Barbosa C, Dierking K (2017) Enhanced behavioral immune defenses in a C. elegans C-type lectin-like domain gene mutant. Dev Comp Immunol 74:237–42
Peleg AY, Tampakakis E, Fuchs BB, Eliopoulos GM, Moellering RC, Mylonakis E (2008) Prokaryote-eukaryote interactions identified by using Caenorhabditis elegans. Proc Natl Acad Sci USA 105(38):14585–90
Petersen C, Dirksen P, Schulenburg H (2015) Why we need more ecology for genetic models such as C. elegans. Trends Genet 31(3):120–7
Pimentel H, Bray NL, Puente S, Melsted P, Pachter L (2017) Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 14(7):687–90
Portal-Celhay C, Blaser MJ (2012) Competition and resilience between founder and introduced bacteria in the Caenorhabditis elegans gut. Infect Immun 80(3):1288–99
Raberg L, de Roode JC, Bell AS, Stamou P, Gray D, Read AF (2006) The role of immune-mediated apparent competition in genetically diverse malaria infections. Am Nat 168(1):41–53
Rafaluk-Mohr C, Ashby B, Dahan DA, King KC (2018) Mutual fitness benefits arise during coevolution in a nematode-defensive microbe model. Evol Lett 2(3):246–56
Ragland SA, Criss AK (2017) From bacterial killing to immune modulation: Recent insights into the functions of lysozyme. PLoS Pathog 13(9):e1006512
Rancès E, Ye YH, Woolfit M, McGraw EA, O’Neill SL (2012) The relative importance of innate immune priming in Wolbachia-mediated dengue interference. PLoS Pathog 8(2):e1002548
Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H et al. (2019) g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47(W1):W191–W198
Raymann K, Shaffer Z, Moran NA (2017) Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLoS Biol 15(3):e2001861
Rossouw W, Korsten L (2017) Cultivable microbiome of fresh white button mushrooms. Lett Appl Microbiol 64(2):164–70
Russell JA, Moran NA (2005) Horizontal transfer of bacterial symbionts: heritability and fitness effects in a novel aphid host. Appl Environ Microbiol 71(12):7987–94
Ryu H, Kim SH, Lee HY, Bai JY, Nam YD, Bae JW et al. (2008) Innate immune homeostasis by the homeobox gene Caudal and commensal-gut mutualism in Drosophila. Science 319:777–82
Sellegounder D, Liu Y, Wibisono P, Chen CH, Leap D, Sun J (2019) Neuronal GPCR NPR-8 regulates C. elegans defense against pathogen infection. Sci Adv 5(11):eaaw4717
Sifri CD, Begun J, Ausubel FM, Calderwood SB (2003) Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect Immun 71(4):2208–17
Singh UB, Malviya D, Wasiullah, Singh S, Pradhan JK, Singh BP et al. (2016) Bio-protective microbial agents from rhizosphere eco-systems trigger plant defense responses provide protection against sheath blight disease in rice (Oryza sativa L.). Microbiol Res 192:300–12
Trevelline BK, Fontaine SS, Hartup BK, Kohl KD (2019) Conservation biology needs a microbial renaissance: a call for the consideration of host-associated microbiota in wildlife management practices. Proc Biol Sci 286(1895):20182448
Ulrich Y, Schmid-Hempel P (2012) Host modulation of parasite competition in multiple infections. Proc Biol Sci 279(1740):2982–9
Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O et al. (2011) The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 334(653):255–8
Varahan S, Iyer VS, Moore WT, Hancock LE (2013) Eep confers lysozyme resistance to enterococcus faecalis via the activation of the extracytoplasmic function sigma factor SigV. J Bacteriol 195(14):3125–34
Visvikis O, Ihuegbu N, Labed SA, Luhachack LG, Alves AF, Wollenberg AC et al. (2014) Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 40(6):896–909
Vorburger C, Ganesanandamoorthy P, Kwiatkowski M (2013) Comparing constitutive and induced costs of symbiont-conferred resistance to parasitoids in aphids. Ecol Evol 3(3):706–13
Wang S, Dos-Santos ALA, Huang W, Liu KC, Oshaghi MA, Wei G et al. (2017) Driving mosquito refractoriness to Plasmodium falciparum with engineered symbiotic bacteria. Science 357(6358):1399–1402
Wilke AB, Marrelli MT (2015) Paratransgenesis: a promising new strategy for mosquito vector control. Parasit Vectors 8:342
Wong D, Bazopoulou D, Pujol N, Tavernarakis J, Ewbank J (2007) Genome-wide investigation reveals pathogen-specific and shared signatures in the response of Caenorhabditis elegans to infection. Genome Biol 8:R194
Acknowledgements
We are grateful to J. Ewbank for comments on the paper, G. Leonard for bioinformatic assistance and four anonymous reviewers for improving the paper. KCK acknowledges funding from the Wellcome Trust (204826/Z/16/Z) and a European Research Council Starting Grant (COEVOPRO 802242).
Author information
Authors and Affiliations
Contributions
SAF and KCK conceived and designed the project; SAF conducted the experiments; GCD compiled the dataset, ran all analyses, and visualised the data; KCK and GCD wrote the paper, with contributions provided by SAF; KCK provided funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Gerald Heckel.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ford, S.A., Drew, G.C. & King, K.C. Immune-mediated competition benefits protective microbes over pathogens in a novel host species. Heredity 129, 327–335 (2022). https://doi.org/10.1038/s41437-022-00569-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00569-3
