
Abstract
Salinity stress adversely affects plant growth and causes considerable losses in cereal crops. Salinity stress tolerance is a complex phenomenon, imparted by the interaction of compounds involved in various biochemical and physiological processes. Conventional breeding for salt stress tolerance has had limited success. However, the availability of molecular marker-based high-density linkage maps in the last two decades boosted genomics-based quantitative trait loci (QTL) mapping and QTL-seq approaches for fine mapping important major QTL for salinity stress tolerance in rice, wheat, and maize. For example, in rice, ‘Saltol’ QTL was successfully introgressed for tolerance to salt stress, particularly at the seedling stage. Transcriptomics, proteomics and metabolomics also offer opportunities to decipher and understand the molecular basis of stress tolerance. The use of proteomics and metabolomics-based metabolite markers can serve as an efficient selection tool as a substitute for phenotype-based selection. This review covers the molecular mechanisms for salinity stress tolerance, recent progress in mapping and introgressing major gene/QTL (genomics), transcriptomics, proteomics, and metabolomics in major cereals, viz., rice, wheat and maize.
Similar content being viewed by others

Introduction
Crop plants experience many biotic and abiotic stresses at different stages during their life cycle. Like other major abiotic stresses (drought and heat), soil salinity threatens global food security, affecting one-quarter to one-third of global crop production (Munns 2002; Abbasi et al. 2015). Soil salinity affects plant growth and development, decreasing agricultural production worldwide (Zhu 2001; Wang et al. 2019; Zhang et al. 2021). More than 800 million hectares of land are affected by salinity, accounting for 6% of the earth’s total land area and 20% of the total cultivated land area (Munns and Tester 2008; Sandhu et al. 2020; Zhang et al. 2021). The saline area is expected to increase due to the application of high salt irrigation water owing to insufficient rainfall and poor agricultural practices (Luo et al. 2017; Zhang et al. 2021), particularly in arid and semi-arid areas with higher evapotranspiration than precipitation (Hanin et al. 2016; Kashyap et al. 2017; Jha et al. 2019). In addition, the climate change-induced rise in mean sea level results in flooding, especially in coastal areas causing soil salinity (Nicholls et al. 2011; Church et al. 2003). Salt-affected land is estimated to reach 50% of the total arable area by 2050 (Wang et al. 2003; Shrivastava and Kumar 2015; Kumar and Sharma 2020), making it challenging to meet the projected food production needs for the increasing human population (Tnay 2019).
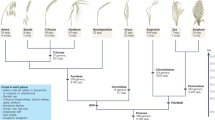
Rice, wheat, and maize are the most important cereal crops, accounting for >50% of the world’s calorie consumption (Gibbon 2012; Luo et al. 2019). Human consumption accounts for 85% of total rice, 72% of total wheat, and 19% of total maize production (http://www.knowledgebank.irri.org). Globally, wheat is ranked first for area planted, maize for production, and rice for production value (Benavente and Giménez 2021). The demand for major cereal crops will increase threefold by 2050 to feed nine billion people. However, salt stress is a major concern for cereal crop production globally as it affects different plant growth stages and leads to abnormalities at the phenotypic, morphological, biochemical, and physiological levels (James et al. 2011). Rice is more sensitive to salt stress than maize (moderately sensitive) and wheat (moderately tolerant). Hence, to address yield losses due to salinity, development of salt-tolerant cultivars (without yield penalty under salt-stressed conditions) is an important requirement (Kashyap et al. 2017; Jha et al. 2019). Conventional breeding has had limited success due to the complex genetic nature of salt tolerance. Recent advances in ‘omics’ technologies, such as genomics, transcriptomics, metabolomics, and QTLomics, can help identify key genes and biomolecules regulating salt tolerance (Ahmad et al. 2016).
Several studies have reviewed various aspects of salt tolerance in different crops (Jha et al. 2019; Gupta et al. 2020; Chen et al. 2021b; Singh et al. 2021), but none have comprehensively integrated the literature on the application of all omics approaches for salt stress tolerance in major cereal crops, viz., rice, wheat, and maize. Here, we review, the impact of salinity on the growth and development of rice, wheat and maize, the adaptive mechanisms involved, and progress in genomic-assisted breeding for salinity stress tolerance in cereal crops. Recent progress in transcriptomics and proteomics is also discussed to reveal the candidate genes and proteins imparting salinity stress tolerance in wheat, maize and rice. The comparative assessment of candidate genes or proteins across the major cereal crops revealed key common genes imparting salinity stress tolerance. Hence, this comprehensive review will serve as a useful guide for salinity breeders to understand the common genes/proteins imparting salinity stress tolerance.
Impact of soil salinity on cereal crops
Soil salinity is defined as a high concentration of solute salts in soils, causing more than 4 dS/m electric conductivity. Salinity adversely affects plant growth and development through its effect on physiological and biochemical pathways (Nabati et al. 2011), severely constraining major cereal crop production, with yield losses of 60% in wheat (Triticum aestivum L.) (El-Hendawy et al. 2017) and 50% in rice (Oryza sativa L.) (Van Genuchten and Gupta 1993), and reductions of 51.43% dry weight and 53.18% leaf area, in maize (Zea mays L.) (Hussein et al. 2007). However, the extent of crop damage varies with salt-stress timing, crop growth stage, plant type and genotype, and climatic conditions (Shahverdi et al. 2018). Each crop has a salt tolerance threshold level measured as the electrical conductivity of a saturated paste of soil (ECe). Rice is the most sensitive crop to salinity (3 dS/m) (Munns and Tester 2008), maize is moderately sensitive (1.8 dS/m) (Rhoades et al. 1992), and wheat is moderately tolerant (6 dS/m). However, the threshold varies depending on the target trait and crop growth stage (Fig. 1b).

Rice is sensitive to salinity particularly at the seedling and panicle initiation to flowering stages (Moradi et al. 2003), whereas wheat is the most vulnerable to salinity during early development (i.e., germination) (Oyiga et al. 2016), hampering water and nutrient uptake, and thus affecting crop growth (Meena et al. 2020). Maize is most vulnerable to salinity stress during germination and seedling establishment; however, at later stages, salinity stress results poor kernel set and reduced grain weight and number, reducing overall yield (Farooq et al. 2015). Under saline conditions, the high concentration of sodium (Na+) ions leads to unbalanced cellular homeostasis, nutrient deficiency, and oxidative stress, affecting growth and causing cell death (Ahanger and Agarwal 2017). Salinity drastically hampers photosynthesis, causing stomatal closure (Munns and Tester 2008), chlorophyll malfunctioning (Jiang et al. 2012), and disrupting the enzymatic machinery of photosynthetic apparatus (Mittal et al. 2012), and chloroplast structure (Quintero et al. 2007; Gengmao et al. 2015). Various studies have revealed that high concentrations of Na+ and Cl– ions in cell sap stimulate a lower osmotic gradient in the nutrient medium, reducing water and nutrient uptake and drastically affecting plant morphological traits (Cantabella et al. 2017; Sapre et al. 2018).
Adaptive mechanisms for salinity stress tolerance
Several physiological, biochemical, and molecular mechanisms are involved in the crop response to salt stress (Munns and Tester 2008, Jha et al. 2019) which can be classified into three main categories (Fig. 2): (1) osmotic tolerance—long-distance signals that reduce shoot growth and positively regulate the production of compatible solutes to maintain leaf expansion and stomatal conductance (Blum 2017); (2) ion exclusion—Na+ transport mechanism in roots that prevents Na+ accumulation reaching to toxic levels in leaves (Munns and Tester 2008); (3) tissue tolerance—high salt concentrations present in leaves but Na+ compartmentalize at the cellular and intracellular level (especially in vacuoles). Osmoprotectant mechanisms include proline and glycine betaine accumulation in maize (Farooq et al. 2015; Iqbal et al. 2020) and reactive oxygen species (ROS) scavenging, glycine betaine accumulation, and hormone modulation in wheat (Shahid et al. 2020). The major genes involved in salt exclusion are salt overlay sensitive (SOS) homologs (OsSOS1, OsCIPK24, and OsCBL4) in rice (Martínez-Atienza et al. 2007), TaSOS1 and TdSOS1 in wheat (Xu et al. 2008; Feki et al. 2011) and ZmNHX7 as a Na+/H+ antiporter in maize (Bosnic et al. 2018). The NHX group of tonoplast-based Na+/H+ exchangers render the vacuolar sequestration of Na+. Overexpression of OsNHX1 and OsVP1 improved salinity tolerance in rice. Similarly, several Na+ sequestration NHX genes were characterized in the vacuoles, including five NHX genes in rice (OsNHX1 to OsNHX5) (Fukuda et al. 2011), four in wheat (TaNHX1, TaNHX2, TaNHX3, and TaNHX4-B) (Brini et al. 2007; Lu et al. 2014; Sharma et al. 2020) and six in maize (ZmNHX1 to ZmNHX6) (Zörb et al. 2005). High-affinity potassium (HAK) transporters also help to induce salt tolerance; 27 HAK genes in rice and maize (Yang et al. 2014, 2020; Zhang et al. 2012), and 56 in wheat were reported (Cheng et al. 2018; Amirbakhtiar et al. 2019). Besides ionic mechanisms, phytohormone-mediated salt tolerance mechanisms are well documented in rice, wheat, and maize, such as abscisic acid, salicylic acid, and jasmonic acid in rice (Yang and Guo 2018; Quilis et al. 2008; Kurotani et al. 2015), wheat (Shakirova et al. 2003) and maize (Iqbal et al. 2020; Ahmad et al. 2019).

Non-selective cation channels (NSCC) and high-affinity potassium (HAK) transporters provide entry points for Na+ (huge influx) and K+ in roots, respectively. The role of voltage insensitive NSCC (VI-NSCC) in Na+ uptake under salt stress has been demonstrated in rye, barley, wheat, and several other plant species. HAK transporters work to balance the augmented Na+ levels by increasing K+uptake (for Na+/K+ homeostasis). Once inside root cells, salt overlay sensitive (SOS) homologs jump in to exclude Na+ from cells. SOS1 is a Na+/H+ antiporter belonging to the cation protein antiporter family with homologs in rice (OsSOS1), wheat (TaSOS1 and TdSOS1), and maize (ZmNHX7). It is primarily involved in Na+ exclusion in roots after activation by serine-threonine kinase (SOS2) and myristoylated calcium-binding protein (SOS3) complex. Na+ accumulated in the cytosol is routed to vacuoles through another set of transporters called Na+/H+ exchangers (NHX), which guide the vacuolar sequestration of Na+. NHX transporters are well characterized in roots and leaves of rice (OsNHX1 to OsNHX5), wheat (TaNHX1toTaNHX3 and TaNHX4-B), and maize (ZmNHX1toZmNHX6). HKT are high-affinity K+ transporters involved in excluding Na+ from xylem vessels and candidate genes for salt-tolerance-related QTL in rice, wheat, and maize. The exclusion of Cl– ions is mediated by a set of transporters—cation chloride co-transporter (CCC) and chloride transporter (CLC) genes. CCC mediates the exclusion of Cl– from roots, while CLC transport Cl– and contributes to salt tolerance.
Genomics-assisted breeding for salinity stress tolerance
Genomics-assisted breeding involves genomic mapping and subsequent introgression to develop improved cultivars. Mapping quantitative trait loci (QTL) is an important approach for understanding the genetic basis of different complex traits governed by multiple genes. Salinity stress tolerance is controlled by polygenes and hence exhibits quantitative inheritance (Jha et al. 2019; Ganie et al. 2019). The availability of molecular markers, mainly microsatellites or simple sequence repeats (SSRs) and, more recently, single nucleotide polymorphism (SNP), facilitate mapping studies. QTL mapping and other mapping approaches, such as association mapping and Bulk-seq/QTL-seq (using extreme, i.e., low and high performing, bulks), have been used extensively to map salt tolerance associated traits, particularly in rice (Prakash et al. 2020).
Mapping major QTL: current status
Numerous minor QTL have been identified using bi-parental mapping populations and association panels, but major QTL are limited (see Tables 1 and 2). Various component traits govern salt tolerance at the seedling stage in rice, particularly root and shoot Na+ and potassium (K+) concentrations, Na+ and K+ uptake, root and shoot length, and leaf chlorophyll concentration (Van Zelm et al. 2020). At the reproductive stage, traits such as root and shoot biomass, grain and straw yield, leaf chlorophyll content, days to flowering, tiller number, and spikelet fertility are typically considered important (Hernández 2019; VanZelm et al. 2020). Several standard visual scoring methods or selection indices (based on key traits), such as the stress susceptibility index (SSI), salt tolerance score (STS), salt injury score (SIS) and salt tolerance ranking (STR) reflect overall plant growth and development, and hence can be used to classify germplasm.
Bulk segregant analysis (BSA)
BSA-based approaches are quick and effective for mapping major QTL, as evident in rice (Michelmore et al. 1991; Choudhary et al. 2019). Tiwari et al. (2016) performed BSA-based mapping using 6068 polymorphic SNPs in CSR11 and MI48 based recombinant inbred line (RIL) populations to identify 21 QTL associated with salt tolerance. Recently, Wu et al. (2020) performed BSA-based QTL mapping to map the loci qST1.1 contributing to salt- tolerance in ‘Sea Rice 86’. Sun et al. (2019) used a QTL-seq approach and two extreme bulks from a segregating population of ‘Changmaogu’ (salt-tolerant) and ‘Zhefu802’ (salt-susceptible) to identify six candidate genes associated with salt tolerance on rice chromosome 1. They identified OsPP2C8 (Os01g0656200) as a candidate gene with sequence polymorphism in ORFs, differentially expressed at the seedling stage. Lei et al. (2020) mapped a major QTL qRSL7 for relative shoot length on chromosome 7 using a QTL-seq approach in an F2:3 population (‘IR36’ × ‘Weiguo’). Furthermore, Lei et al. (2020) identified a candidate gene Os07g0569700 (OsSAP16) using an RNA-seq and sequence variation approach for salt tolerance. In wheat, Shamaya et al. (2017) conducted BSA using 9K SNP markers to map two major loci for third leaf Na+ concentration on chromosomes 3B and 4B, with the gene on 4B likely to be a high-affinity K+ transporter (HKT1;5-B1).
QTL mapping: Classical approach
Rice
QTL mapping for salt tolerance at the seedling and reproductive stages in rice used mapping populations derived from a cross between salt-tolerant and salt-sensitive genotypes (Singh et al. 2007). Several salt-tolerant rice genotypes, including ‘Nona Bokra’ (Puram et al. 2018), ‘Jiucaiqing’ (Wang et al. 2012), ‘CSR27’ (Pandit et al. 2010), ‘Pokkali’ (Chen et al. 2019), ‘Gharib’ (Ghomi et al. 2013), ‘Changbai10’ (Zheng et al. 2015), ‘Capsule’ (Rahman et al. 2019), ‘Changmaogu’ (Sun et al. 2019), ‘Kalajoha’ (Mazumder et al. 2020), ‘Sea Rice 86’ (Wu et al. 2020) and ‘Hasawi’ (Rahman et al. 2017), have been used to map traits associated with seedling stage salt tolerance (Table 1).
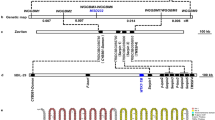
‘Hasawi’ (Bimpong et al. 2014), ‘Pokkali’ (Khan et al. 2015), and ‘CSR10’ (Pundir et al. 2021) were used as the tolerant parent for mapping salt-tolerance associated traits at the reproductive stage. ‘Pokkali’ is salt-tolerant at the seedling and reproductive stage (Table 1). QTL have been identified in the mapping populations using QTL mapping or a whole-genome resequencing-based QTL-seq approach (Krishnamurthy et al. 2020). In rice, ‘Saltol’ on chromosome 1 is the most commonly occurring QTL governing salt tolerance at the seedling stage (Babu et al. 2017b). The ‘Saltol’ genomic region has been well characterized in several landraces and genotypes (Manohara et al. 2021). A major QTL, qSSISFH8.1, on chromosome 8 (flanked by HvSSR08-25 and RM3395) governing SSI for spikelet fertility under salinity stress at the reproductive stage can be used to impart salt tolerance (Pandit et al. 2010; Pundir et al. 2021). Identified QTL on chromosomes 2, 3, 4, 9, 10, and 11 at the seedling and reproductive stage indicate that these chromosomes are relatively more important for harboring genes for seedling and reproductive stage salinity tolerance in rice (Table 1).
Wheat
QTL mapping efforts in wheat identified >500 QTL (distributed over 21 chromosomes) for salinity tolerance associated traits, with a few dozen major QTL explaining more than 10% of the phenotypic variance (Gupta et al. 2020; Singh et al. 2021). Since 2011, 16 QTL mapping studies have been published, most focusing on salt tolerance at the seedling stage (Table 1). Hussain et al. (2017) reported a major QTL, qSNAX.7A.3, for shoot dry weight, a commonly used direct measure of salinity stress tolerance. Asif et al. (2018) detected three major QTL, QG(1–5), asl-7A (relative growth rate), QNa.asl-7A (leaf Na+ concentration), and QK.asl-5A (leaf K+ concentration), in an ‘Excalibur’ × ‘Kukri’ doubled haploid (DH) population. QK.asl-5A was in the vicinity of the vernalization response gene (Vrn-A1), but QG(1–5).asl-7A was the prominent QTL, present in six of 44 Australian bread and durum wheat cultivars. Devi et al. (2019) identified 25 QTL under sodic conditions using RILs derived from ‘HD2009’ and ‘Kharchia 65’. The study revealed the linkage of SSR marker gwm 26 for K+ content (QSK+.iiwbr-2D) and cfd 84 for tiller number (QStn.iiwbr-4D) and earhead number (QSne.iiwbr-4D). Ilyas et al. (2020) mapped 60 QTL; four (for total chlorophyll, water potential, and Na+ content) were located on chromosome 6B in the vicinity of gwm70 and Xbarc361 markers; hence, chromosome 6B is a good source of salinity stress tolerance in wheat. Recently, Luo et al. (2021) used a wheat 55K SNP array to map 90 stable QTL for 15 agronomic traits in a ‘Zhongmai 175’ × ‘Xiaoyan 60’ RIL population screened under low and high levels of salt stress at the adult stage. Eight QTL from four QTL clusters were validated in natural populations, and competitive allele-specific PCR (KASP) markers were designed for three QTL clusters. Hence, many major QTL are now available in wheat (more than maize but fewer than rice) (Table 1). In wheat, chromosomes 3A, 5B, and 6B possess QTL for salt-tolerance-associated traits at the seedling and adult stage (Table 1), which are relatively anticipated to harbor multi-stage tolerance.
Maize
There are limited QTL mapping studies on salinity stress tolerance in maize (Table 1). Two major QTL, QFgr1 and QFstr1, for field germination rate and field tolerance ranking, respectively, were mapped on chromosome 1 using ‘F63’ × ‘F35’ based RILs phenotyped under saline field conditions. Nine conditional QTL for shoot fresh weight, tissue water content, shoot Na+ concentration, shoot K+ concentration, and shoot K+/Na+ ratio were mapped on chromosomes 1, 3, 4, and 5 (Cui et al. 2015b). Luo et al. (2017) identified a major QTL for plant height, qSPH1 (with phenotypic variance of 31%), on chromosome 1 under saline conditions. The study also identified a major QTL for a plant-height-based salt tolerance index on the same chromosome and two candidate genes, GRMZM2G007555 and GRMZM2G098494, that code for ion homeostasis regulation likely associated with salt tolerance. A major QTL, ZmNC1 conferring leaf Na+ concentration, was reported on chromosome 3 in RILs derived from crossing ‘Zheng58’ and ‘Chang7-2’ (Zhang et al. 2018). The authors also performed transcriptomic analysis to identify a plasma-membrane-localized class I HKT ion transporter (ZmHKT1) as a putative candidate gene for ZmNC1. Later, 65 QTL were mapped for biomass-related traits under salinity stress, with 13 major QTL (for nine salt-tolerance-related traits) on chromosome 1, explaining more than 21% of the phenotypic variation (Luo et al. 2019). Hence, the 17 QTL identified on chromosome 1 can be targeted for introgression to develop salt-tolerant maize cultivars.
Association mapping to identify candidate genes for salinity stress tolerance
The exploitation of historical recombination events through association mapping (AM) or genome-wide association studies (GWAS) has been instrumental in identifying genomic regions responsible for specific traits in various crop species, and salt tolerance is not an exception (Table 2). GWAS has been used widely in cereal crops (Gupta et al. 2014) to identify genes related to abiotic stress tolerance (Kumar et al. 2015; Frouin et al. 2018; Gupta et al. 2020). More GWAS studies for salinity stress tolerance have been conducted in rice than wheat or maize, as rice is the most salt-sensitive (Table 2).
Rice
In an AM panel of 220 rice accessions, mostly indica type, Kumar et al. (2015) identified novel salt-tolerance-related marker-trait associations (MTAs) on chromosomes 1, 4, 6, and 7. Another AM panel of 208 rice accessions from the mini core collection was used (Naveed et al. 2018) to identify 20 MTAs associated with 11 salt-tolerance-related traits. Mild seedling stage salinity stress tolerance was used to map associated genes in a panel of 235 temperate japonica rice accessions (Frouin et al. 2018). After the release of the 3K rice genome sequence, Batayeva et al. (2018) used 203 temperate japonica rice accessions to identify 26 MTAs for nine salt-tolerance-related traits at the seedling stage. Most of the QTL were located close to the genes governing kinase and calcium signaling and metabolism. In another study using a large panel of 708 rice accessions, five known (OsSUT1, OsMYB6, OsHKT1;4, OsGAMYB, and OsCTR3) and two novel (LOC_Os02g49700 and LOC_Os03g28300) genes were significantly associated with yield and related traits under salinity stress at the seedling stage (Liu et al. 2019). A panel of 104 Thai rice accessions of indica rice was evaluated to identify candidate genes related to salinity stress tolerance at the flowering stage; the genes were mainly distributed on chromosomes 1, 2, 8, and 12 (Lekklar et al. 2019). A study using a large panel of 664 accessions from the 3K Rice Genome Project and high-density genotypic data of 17 million SNPs identified 21 QTL with two candidate genes (OsSTL1 and OsSTL2) confirmed via sequence analysis (Yuan et al. 2020). An evaluation of accessions under hydroponic and soil media identified 97 and 74 QTL, respectively, with 11 QTL common to both (Chen et al. 2020). The most significant QTL on chromosome 1 harbored two post-translational modification genes, OsSUMO1 and OsSUMO2. Warraich et al. (2020) identified genomic locations responsible for several salt-tolerance-associated traits, including Na+, K+, Ca2+, and Mg2+ content, in leaves and stems at the reproductive stage. Recently, Nayyeripasand et al. (2021) identified 29 genomic regions for salinity stress tolerance in a panel of 155 rice varieties, including two and three novel candidate genes on chromosomes 8 and 1, respectively. The above GWAS studies involved different mapping panels, which should help better understand the mechanisms of salinity stress tolerance in rice.
Wheat
Using natural recombination events, a panel of 150 wheat accessions was genotyped with the 90K SNP chip identified 37 QTL for salt-tolerance-related traits at three growth stages, i.e., germination, seedling, and adult (Oyiga et al. 2018). In another mapping panel of 100 bread wheat, seven SNPs were associated with genes (four candidate genes) linked to Na+ accumulation/exclusion. Hu et al. (2021) conducted GWAS at the adult plant stage using a panel of 191 accessions to establish 11 QTL for salt-tolerance-related traits. Significant SNPs were validated using two RIL populations.
Maize
Fewer studies on salinity stress tolerance have been conducted in maize than rice or wheat (Table 2). In a candidate-gene-based AM approach, a panel of 54 diverse Chinese inbred lines was used to find associations with sequence variation (two SNPs within the coding region) in the ZmHKT1-5 gene for salinity stress tolerance (Jiang et al. 2018). In another study, 57 SNPs had significant associations with early-vigor-related traits, with 40 associated with shoot-biomass-related traits (Sandhu et al. 2020). Cao et al. (2020) genotyped a panel of 419 inbred lines with 55K chip-based SNPs to reveal two closely located SNPs significantly associated with shoot Na+ content (candidate gene ZmNSA1 conferring shoot Na+ content variation). Recently, Liang et al. (2021) conducted metabolic GWAS to identify 37 metabolite biomarkers and 10 candidate genes for salt-induced osmotic stress tolerance.
Genomic selection for salinity stress tolerance in cereals
Genomic selection (GS) is an efficient tool for assessing the breeding value of a genotype based on its sequence information (Goddard and Hayes 2007). It is a practical breeding tool for selecting superior genotypes among the available diversity and is a better solution than marker-assisted selection (MAS), especially for complex traits such as salinity stress tolerance (governed by several genes and metabolic pathways), due to the inclusion of minor genes for determining the worth of the genotype. As far as rice, wheat and maize are concerned, there have been no systematic GS efforts for salinity stress tolerance. However, a haplotype-based GS approach can be used to charter customized varieties suitable for a particular region with a specific stress type and consumer quality preferences (Sinha et al. 2020). The publicly available 3K rice genome sequence information can be used to predict haplotypes suitable for salinity stress and design a breeding strategy.
Transcriptomics for salinity tolerance
Advances in high-throughput methods for next-generation sequencing offer a better understanding of salinity stress tolerance in crops by identifying salt-responsive genes using genomics and transcriptomics (Egan et al. 2012; Duarte-Delgado et al. 2020; Kashyap et al. 2020). Transcriptomics can identify stress-associated essential transcripts, the transcriptional structure of genes, their functional pathways, and other post-transcriptional modifications (Wang et al. 2009). RNA-seq and microarray are gene expression approaches (Table 3) with the former being more popular due to its precise transcript measurement ability (Wang et al. 2009).
Rice
Kumari et al. (2009) first reported 1,194 salinity-regulated cDNA between two contrasting genotypes, ‘IR64’ and ‘Pokkali’. The latter exhibited enhanced gene expression in the GST, LEA, CaMBP V-ATPase, and OSAP1 zinc finger protein families. Later, in transcriptomic studies involving ‘IR64’ and ‘Pokkali’ under salt stress, Shankar et al. (2016) reported that the upregulation of transcripts was involved in wax and terpenoid metabolism. Whole-genome resequencing and transcriptome analysis of ‘Sea Rice 86’ identified several candidate genes for salt adaptation (Chen et al. 2017). Li et al. (2018) concluded that ‘Pokkali’ (tolerant) has more stable mRNA and can stably load mRNA on polysomes during salt stress than ‘IR29’ (sensitive). Chandran et al. (2019) used RNA-seq based transcriptomic analysis in japonica rice cultivar ‘Chilbounder’ screened under stress and non-stress conditions to identify 447 differentially expressed genes (DEGs), which were mainly involved in carbohydrate and amino-acid metabolic processes. Recently, Xie et al. (2021) reported that the AP2/EREBP-HB-WRKY cascade is activated by stress, imparting melatonin-mediated salt tolerance in rice.
Wheat
Numerous transcriptomics studies have been conducted in wheat using different plant parts (roots, shoots, both or total plant) to understand salt stress tolerance (Table 3). Diacylglycerol kinase encoding gene, signal transduction module gene, and GST were upregulated for stress tolerance in wheat (Liu et al. 2012). Under salt stress, the salt-stress tolerant gene, TaMYB73, was upregulated in roots but downregulated in leaves. However, overexpression of the gene in Arabidopsis improved stress tolerance (He et al. 2012). Mahajan et al. (2020) found that root-growth-enhancing genes encoding expansin, xyloglucan endotransglucosylase/hydrolase, dehydrins, and peroxidases improve salt stress tolerance. Additionally, the study identified 10,805 unigenes in ‘Kharchia Local’ roots at anthesis, and WRKY, MYB, NAC, bHLH, AP2/ERF transcription factors (TFs) were upregulated during salt stress. For the first time, Amirbakhtiar et al. (2019) identified 26,171 novel transcripts from the roots of ‘Chinese Spring’ seedlings using RNA-seq. Under stress conditions, Ta.ABAC15, an ABC transporter gene, was significantly upregulated. Cytosolic-calcium-level controlling genes Ta.ANN4, Ta.ACA7, and Ta.NCL2,29 NAC genes, and 48 MYB TFs also increased under salt stress. Besides differentially expressed TFs, miRNAs also contribute to stress tolerance, regulating transcription and translation. In wheat roots, 16 novel salt-stress-associated miRNAs were identified. Stress-responsive miRNAs, athmiR5655, osamiR172b and osamiR444b.2, regulated TFs, bHLH135like, AP2/ERBP, and MADS box, respectively (Eren et al. 2015). Duarte-Delgado et al. (2020) found 50 calcium-binding genes and 18 xyloglucans:xyloglucosyl transferase (cell wall genes) that reduce osmotic stress; subgenome D had the most (35.8 ± 1.7%) salt-responsive genes, with TraesCS2A02G395000 the strongest salt-responsive gene. Ma et al. (2020) reported the upregulation of TaCYP450 and six ethylene-dependent genes during salt stress. Upregulation of genes associated with LEA, dehydrin, and potassium transportation, and sodium/cation exchange and aquaporin in roots and shoots, respectively, enhanced salt stress tolerance (Bhanbhro et al. 2020). A reduction in ethylene sensitivity enhances salt tolerance. Upregulation of nine novel ETHYLENE RESPONSE FACTORs (ERFs) (TaERF1, 2, 3, 4, and 6) reduced ethylene sensitivity and improved salt tolerance in wheat (Ma et al. 2021).
Maize
In maize, a reasonable number of transcriptomics studies are available (Table 3). Using RNA-seq analysis, Li et al. (2017) reported 1661 DEGs between the salt stress and control treatments at the seedling stage, which were associated with hormone metabolism, signaling, TFs, fatty acid biosynthesis, and lipid signaling. Chen et al. (2020) reported 219 upregulated and 153 downregulated DEGs in salt-sensitive and tolerant lines, respectively, which were associated with ion homeostasis, strong signal transduction activation, and increased ROS scavenging and different TFs (MYB, MYB-related, AP2-EREBP, bHLH, and NAC families). Similarly, Zhang et al. (2021) identified 459 DEGs, of which Aux/IAA, SAUR, and CBL-interacting kinase reportedly regulate salt tolerance. In addition, WRKY, bZIP, and MYB TFs act as regulators in the salt-responsive regulatory network of maize roots. Comparing transcriptomics studies in wheat, maize, and rice revealed some common TF families, such as WRKY, MYB, AP2/ERF, and NAC, important for imparting salinity stress tolerance (Table 3). These TFs maintain cellular homeostasis and osmotic balance in plants by regulating the expression of salt-stress responsive genes and need to be explored to understand their specific modes of action.
Proteomics for salinity stress tolerance
Proteomics helps to capture insights into salt-stress tolerance by identifying protein modulations and pathway modifications (Parvaiz et al. 2016). Proteomics/metabolomics studies for salt tolerance have been mostly conducted in wheat followed by maize and rice (Table 4).
Rice
Nam et al. (2015) performed metabolic profiling of 38 rice genotypes using H-NMR spectroscopy and found glutamine and allantoin positively correlated with salt tolerance. Wang et al. (2021) undertook metabolic profiling of rice genotypes to confirm the role of several metabolites, including amino acids (valine and proline) and organic acids (phosphoenolpyruvic acid, glyceric acid, ascorbic acid), in imparting salt tolerance.
Wheat
Phosphoproteomics identified the salt-stress response and defense biomarkers [cp31BHv, betaine-aldehyde dehydrogenase (BADH), cytosolic (GS1), Cu/Zn superoxide dismutase (SOD), MAT3, leucine aminopeptidase 2 (LAP2), and 2-Cys peroxiredoxin BAS1] in wheat (Lv et al. 2016). Cu/Zn SODs, GSTs, DHNs, and V-ATPase were upregulated in roots, and Cu/Zn SODs, LEA, and DHN in leaves (Lv et al. 2016). Jiang et al. (2017) analyzed wheat seedling roots and reported three salt-tolerance-associated proteins: a pyruvate orthophosphate dikinase (PPDK) and two late embryogenesis-abundant (LEA) encoded by TaPPDK, TaLEA1, and TaLEA2, respectively. Ubiquitination-related and pathogen-related proteins, membrane intrinsic protein transporters, TFs, and antioxidant enzymes imparted salt-stress tolerance through single cellular homeostasis in roots. Protecting cell structure during salt stress is an important strategy for stress tolerance. Singh et al. (2017) reported the upregulation of tubulin, profilin, retinoblastoma, casparian strip membrane protein, xyloglucan endotransglycosylase, and ion transporter proteins (e.g., malate transporter) for enhanced salt tolerance. Three genes, TraesCS6A01G336500.1, TraesCS4B01G254300.1, and TaABCF3, encoding OPAQUE1, NRAMP-2, and transporter genes, respectively, enhanced salt stress tolerance by balancing the shoot Na+/K+ ratio, specific energy fluxes for absorption, dissipation, and shoot Na+ uptake (Oyiga et al. 2019). Han et al. (2019) reported the upregulation of five SODs, three malate dehydrogenases and dehydrin proteins, and one V-ATPase protein in roots, and two Cu/Zn, one LEA protein, and DHN proteins in leaves under salt stress. Ethylene modifies or activates ribosomal proteins (RPs) in wheat, reducing ROS accumulation and improving stress tolerance. Eight and 49 differentially expressed proteins (DEPs) were found in roots and shoots, respectively, and 48 RPs in roots (Ma et al. 2020). In chloroplasts of wheat seedling leaves, 117 upregulated differentially accumulated proteins (DAPs) were associated with the Calvin cycle, amino-acid metabolism, carbon and nitrogen metabolism, transcription and translation, and antioxidation (Zhu et al. 2021).
Maize
Protein analysis of ‘F63’ (tolerant) and ‘F35’ (sensitive) maize genotypes using the iTRAQ approach identified 28 salt-responsive proteins, of which 22 were expressed explicitly in ‘F63’ (Cui et al. 2015a). Similarly, Soares et al. (2018) reported 1747 proteins, of which 209 were abundant in response to salt stress and mainly associated with oxidative stress, dehydration, respiration, and translation. Chen et al. (2019) identified 1056 DEPs, of which 626 and 473 were specific to tolerant and sensitive maize inbred lines, respectively. DEPs expressed in the tolerant lines under salt stress were associated with phenylpropanoid biosynthesis, starch and sucrose metabolism, and the MAPK signaling pathway, while those in the sensitive lines were related to the nitrogen metabolism pathway only (Chen et al. 2019). Hence, under salt stress, glucose metabolism is mainly induced in salt-tolerant lines, while nucleic acid metabolism is induced in salt-sensitive lines at the seedling stage.
Comparing proteomics studies across rice, wheat, and maize highlights glutamine as a common, key amino acid (via upregulation of glutamine synthetase) for imparting salinity stress tolerance in salt-tolerant lines (Table 4). Glutamine synthetase enhances salt tolerance in the tolerant cultivars by imparting better photorespiration capacity (Kamal et al. 2012; Nam et al. 2015; Luo et al. 2018). Studies have shown that enhanced proline in rice and wheat, and RPs in wheat and maize impart salt tolerance (Table 4). Hence, these key amino acids can be used as metabolite markers in salinity stress breeding programs in cereal crops.
Meta-analysis for salinity stress tolerance
With the availability of numerous QTL and transcriptomics studies, meta-analysis is the best approach for identifying or predicting consensus genomic regions (candidate genes and meta-QTL i.e., genomic intervals containing two or more QTL from independent studies) for subsequent use in plant breeding programs (Sinha et al. 2021). Meta-analysis can be performed with expression datasets (microarray or RNA-seq) and QTL datasets. For example, Kaur et al. (2016) performed a meta-analysis using a publicly available microarray data set and RNA-seq data to identify 31 candidate genes associated with salt tolerance in rice. Later, Buti et al. (2019) performed a meta-analysis of RNA-seq data in rice to identify chilling, osmotic and salt stress tolerant genes. Later, Islam et al. (2019) predicted 11 meta-QTL for SIS and shoot Na+ and K+ concentrations and identified related candidate genes (using gene expression studies and gene ontology prediction). Kong et al. (2019) performed a meta-analysis of the salt stress transcriptomic response in rice (based on 96 publicly available microarray datasets) and identified 5559 DEGs, with most related to the mitogen-activated protein kinase (MAPK) cascade, Ca2+ signal transduction, hormone signals, TFs and regulators. Recently, Mansuri et al. (2020) used published transcriptomic and QTL mapping studies to predict 3449 DEGs in 46 meta-QTL, with most involved in functions of ion transport, cell wall organization, transcriptional regulation, cell response to stress, transporter activity, TFs, and oxidoreductase.
Epigenomics and genome editing approaches for salt tolerance: recent progress
Recent progress in the technological advances of epigenetics and transgenics is promising for delivering climate-resilient genotypes. The epigenetic changes include chromatin modification, several post-transcriptional histone modifications and DNA methylation which play an important role in imparting salt stress tolerance in crops. The hypermethylation of cytosines at HKT genes in shoots and roots imparted salt tolerance in wheat (Kumar et al. 2017). Wang et al. (2011) reported altered DNA methylation in salt-sensitive and salt-tolerant rice genotypes. Subsequently, methylation-sensitive amplification polymorphism analysis revealed the role of DNA methylation for salt tolerance in rice, evident from enhanced DNA methylation and differential gene expression under high salinity conditions (Karan et al. 2012). Similarly, overexpression of miR156 reduced the expression of TFs (SPL9 and DFR), imparting salt tolerance in rice (Cui et al. 2014). Furthermore, high-throughput sequencing (MeDIP-seq) technologies resolved the differentially methylated regions at the whole-genome level in salt-tolerant rice (Ferreira et al. 2015) and maize (Sun et al. 2018). Recently, Rajkumar et al. (2020) reported cultivar-specific DNA methylation with hypermethylation prominent in salt-tolerant rice cultivar ‘Pokkali’ and hypomethylation in drought-tolerant cultivar ‘Nagina22’ under salt stress. These recent findings indicate the potential role of epigenetic factors in imparting salinity tolerance in rice, wheat and maize.
In recent years, advancements in CRISPR/Cas technology have provided a new avenue for genome editing, enabling target site-editing of desirable alleles to produce desirable phenotypes (Kumar et al. 2020). Salinity tolerance in rice was induced by editing the OsRR22 gene using CRISPR-Cas9 (Zhang et al. 2019). Kumar et al. (2020) used CRISPR-Cas9 based gene editing to produce a mutant Cas9-freedst Δ184–305 in indica rice cultivar ‘MTU1010’, which showed a moderate level of osmotic stress under seedling-stage salt stress. Further, several transgenes were used to induce salinity tolerance in rice for salt exclusion and Na+ compartmentation viz., PpENA1, PyKPA1, SOD2, AtHKT1;1, HmHKT1;5, HvHKT1;5, OsVP1, OsNHX1, CgNHX1, AdNHX1, OsNHX1, PgNHX1 and AtNHX1 (Kotula et al. 2020). The use of genome editing for salt stress tolerance is limited but has the potential for developing salt-tolerant cereal crops.
Genomics-assisted bred salt-tolerant cultivars: achievements
Crop responses to salinity and their tolerance to salinity are complex genetic and physiological phenomena. Only major QTL can be targeted through marker-assisted backcrossing (MABC) as evident in rice (Krishnamurthy et al. 2020). Maize and wheat lack introgression studies for major QTL, which may be due to complex genome size or lack of validation of functionality of identified QTL. However, one successful example is ‘Line149’ in wheat (Munns et al. 2012) developed through introgressing TmHKT1;5-A, a gene from Triticum monococcum that confers Na+ exclusion. Dominant markers, gwm410 and gwm291 and co-dominant marker, cslinkNax2 were used to select favorable lines (Munns et al. 2012). In contrast, there have been remarkable achievements in introgressing major QTL for salinity stress tolerance in rice. ‘Saltol’ is the most celebrated and widely used genomic region for seedling-stage salt tolerance, adapted from ‘FL478’ (derived from local landrace ‘Pokkali’ from Kerala, India) (Bonilla et al. 2002). Fine mapping of this region revealed a major gene, SKC1, which stabilizes K+ homeostasis in the salt-tolerant variety (Ren et al. 2005). SKC1 encodes an HKT-type transporter that is Na+ selective, and maintains K+/Na+ homeostasis under salt stress. ‘FL478’ was used as a universal donor for ‘Saltol’ QTL in the MABC program to improve seedling-stage salt tolerance in several commercial cultivars (Krishnamurthy et al. 2020). In India, several ‘Saltol’ introgression lines, (i.e., near-isogenic lines) have been developed in popular released cultivars, including ‘Sarjoo52’, ‘Pusa Basmati-1’, ‘Pusa Basmati 1121’, and ‘Pusa 1509’, with yields on par with the original variety under non-stressed conditions and better in salinity prone areas (Krishnamurthy et al. 2020; Singh et al. 2018; Babu et al. 2017a). Rana et al. (2019) used the hitomebore salt-tolerant-1 (hst1) gene from an EMS mutant line governing the seedling and reproductive stage to develop the salt-tolerant variety, ‘Kaijin’, which was used to introgress the hst1 gene in ‘Yukinko-mai’ to develop ‘YNU31-2-4’, a salt-tolerant cultivar. The Hst1 gene encodes a B-type response regulator called OsRR22 (Os06g0183100). A substitution mutation in the third exon of this gene governs salt tolerance in this genotype. Marker-assisted recurrent selection (MARS) was recently used to improve drought and salt tolerance in the ‘IR58025B’ genetic male sterile maintainer line, the most common maintainer line used for hybrid rice development in India (Suryendra et al. 2020). The complete list of improved rice lines/cultivars using genomics-assisted breeding is presented in Table 5. The physiological mechanisms of salt tolerance at the reproductive stage in rice differ from those at the seedling stage; hence, a separate breeding strategy should be followed (Pundir et al. 2021; Singh et al. 2021).
Conclusion and future perspectives
Salinity is a major abiotic stress that adversely affects crop growth and development, resulting in yield losses. The reclamation of saline ecologies is a time-consuming and challenging task. Developing salt-tolerant cultivars of rice, wheat, and maize, the staple food crops, is a sustainable and cost-effective approach to safeguard food security. The availability of molecular markers, modern breeding tools, and omics techniques can help to understand the genetic basis of salt tolerance by identifying relevant genomic regions for the quick development of tolerant cultivars instead of the time-consuming conventional breeding approach. A systems biology approach that integrates all omics approaches (genomics, transcriptomics, proteomics, metabolomics and phenomics) is necessary to understand salt-stress tolerance mechanisms in different crops. However, a pre-breeding approach (using wild relatives and landraces) is needed to broaden the genetic base of germplasm for traits associated with salinity stress tolerance, as most crop breeding programs are based on a few donor lines. Rice has witnessed remarkable success, particularly with the Saltol QTL. Similar success is expected in wheat and maize considering the availability of major QTL and emphasis on salinity stress studies. The availability of multi-parent mapping populations in these crops will help identify novel alleles for salinity stress tolerance (Gireesh et al. 2021). Using molecular breeding approaches combined with omics and rapid generation techniques, such as speed breeding to shorten generations and genome editing to edit the target gene, will further hasten the development of salt-tolerant cultivars (Watson et al. 2018; Kumar et al. 2020). Recently, rice witnessed the use of CRISP-Cas for salinity improvement by targeting the OsRR22 and OsDST genes; wheat and maize will likely witness the success of genome-edited cultivars in the near future. Using a pangenomics approach and wild relatives will help identify the core and unique genes for salinity stress adaptation in rice, wheat, and maize (Khan et al. 2020). Identifying cross-tolerance for multiple abiotic stresses and stress memory development is another worth exploring area to develop multi-stress tolerant cultivars (Choudhary et al. 2021). Considering the worldwide salinity crisis, better policies, and allocation of grants and funding for salinity stress research will assist in the sustainable development of salt-tolerant elite cultivars.
References
Abbasi GH, Akhtar J, Ahmad R, Jamil M, Anwar-Ul-Haq M, Ali S et al. (2015) Potassium application mitigates salt stress differentially at different growth stages in tolerant and sensitive maize hybrids. Plant Growth Regul 76:111–125
Ahanger MA, Agarwal RM (2017) Salinity stress induced alterations in antioxidant metabolism and nitrogen assimilation in wheat (Triticum aestivum L) as influenced by potassium supplementation. Plant Physiol Biochem 115:449–460
Ahmad P, Abdel Latef AA, Rasool S, Akram NA, Ashraf M, Gucel S (2016) Role of proteomics in crop stress tolerance. Front Plant Sci 7:1336
Ahmad RM, Cheng C, Sheng J, Wang W, Ren H, Aslam M et al. (2019) Interruption of jasmonic acid biosynthesis causes differential responses in the roots and shoots of maize seedlings against salt stress. Int J Mol Sci 20(24):6202
Amin AY, Diab AA (2013) QTL mapping of wheat (Triticum aestivum L.) in response to salt stress. Int J Bio Technol Res 3:47–60
Amirbakhtiar N, Ismaili A, Ghaffari MR, Nazarian Firouzabadi F, Shobbar ZS (2019) Transcriptome response of roots to salt stress in a salinity-tolerant bread wheat cultivar. PLoS One 14:e0213305
Asif MA, Schilling RK, Tilbrook J, Brien C, Dowling K, Rabie H et al. (2018) Mapping of novel salt tolerance QTL in an Excalibur × Kukri doubled haploid wheat population. Theor Appl Genet 131:2179–96
Azadi A, Mardi M, Hervan EM, Mohammadi SA, Moradi F, Tabatabaee MT et al. (2015) QTL mapping of yield and yield components under normal and salt-stress conditions in bread wheat (Triticum aestivum L.). Plant Mol Biol Rep 33:102–120
Babu NN, Vinod KK, Krishnamurthy SL, Krishnan SG, Yadav A, Bhowmick PK et al. (2017b) Microsatellite based linkage disequilibrium analyses reveal Saltol haplotype fragmentation and identify novel QTLs for seedling stage salinity tolerance in rice (Oryza sativa L.). J Plant Biochem Biotechnol 26:310–320
Babu NN, Krishnan SG, Vinod KK, Krishnamurthy SL, Singh VK, Singh MP et al. (2017a) Marker aided incorporation of Saltol, a major QTL associated with seedling-stage salt tolerance, into Oryza sativa ‘Pusa basmati 1121’. Front Plant Sci 8:41
Batayeva D, Labaco B, Ye C, Li X, Usenbekov B, Rysbekova A et al. (2018) Genome-wide association study of seedling stage salinity tolerance in temperate japonica rice germplasm. BMC Genet 19:1–11
Benavente E, Giménez E (2021) Modern approaches for the genetic improvement of rice, wheat and maize for abiotic constraints-related traits: a comparative overview. Agronomy 11:376
Bhanbhro N, Xiao B, Han L, Lu H, Wang H, Yang C (2020) Adaptive strategy of allohexaploid wheat to long-term salinity stress. BMC Plant Biol 20:1–14
Bimpong IK, Manneh B, Diop B, Ghislain K, Sow A, Amoah NKA et al. (2014) New quantitative trait loci for enhancing adaptation to salinity in rice from Hasawi, a Saudi landrace into three African cultivars at the reproductive stage. Euphytica 200:45–60
Bimpong IK, Manneh B, Sock M, Diaw F, Amoah NKA, Ismail AM et al. (2016) Improving salt tolerance of lowland rice cultivar ‘Rassi’ through marker-aided backcross breeding in West Africa. Plant Sci 242:288–299
Bizimana JB, Luzi-Kihupi A, Murori RW, Singh RK (2017) Identification of quantitative trait loci for salinity tolerance in rice (Oryza sativa L.) using IR29/Hasawi mapping population. J Genet 96:571–582
Blum A (2017) Osmotic adjustment is a prime drought stress adaptive engine in support of plant production. Plant Cell Environ 40:4–10
Bonilla P, Dvorak J, Mackell D, Deal K, Gregorio G (2002) RFLP and SSLP mapping of salinity tolerance genes in chromosome 1 of rice (Oryza sativa L.) using recombinant inbred lines. Philipp Agric Scientist 85:68–76
Borrelli GM, Fragasso M, Nigro F, Platani C, Papa R, Beleggia R et al. (2018) Analysis of metabolic and mineral changes in response to salt stress in durum wheat (Triticum turgidum ssp. durum) genotypes, which differ in salinity tolerance. Plant Physiol Biochem 1:57–70
Bosnic P, Bosnic D, Jasnic J, Nikolic M (2018) Silicon mediates sodium transport and partitioning in maize under moderate salt stress. Environ Exp Bot 155:681–687
Brini F, Hanin M, Mezghani I, Berkowitz GA, Masmou K (2007) Overexpression of wheat Na+/H+ antiporter TNHX1 and H+-pyrophosphatase TVP1 improve salt- and drought-stress tolerance in Arabidopsis thaliana plants. J Exp Bot 58:301–308
Buti M, Baldoni E, Formentin E, Milc J, Frugis G, Lo Schiavo F et al. (2019) A meta-analysis of comparative transcriptomic data reveals a set of key genes involved in the tolerance to abiotic stresses in rice. Int J Mol Sci 20:5662
Cantabella D, Piqueras A, Acosta-Motos JR, Bernal-Vicente A, Hernández JA, Díaz-Vivancos P (2017) Salt-tolerance mechanisms induced in Stevia rebaudiana Bertoni: Effects on mineral nutrition, antioxidative metabolism and steviol glycoside content. Plant Physiol Biochem 115:484–496
Cao Y, Zhang M, Liang X, Li F, Shi Y, Yang X et al. (2020) Natural variation of an EF-hand Ca2+-binding-protein-coding gene confers saline-alkaline tolerance in maize. Nat Commun 11:1–14
Capriotti AL, Borrelli GM, Colapicchioni V, Papa R, Piovesana S, Samperi R et al. (2014) Proteomic study of a tolerant genotype of durum wheat under salt-stress conditions. Anal Bioanal Chem 406:1423–35
Chandran AKN, Kim JW, Yoo YH, Park HL, Kim YJ, Cho MH et al. (2019) Transcriptome analysis of rice-seedling roots under soil–salt stress using RNA-Seq method. Plant Biotech Rep 13:567–578
Chen C, Norton GJ, Price AH (2020) Genome-wide association mapping for salt tolerance of rice seedlings grown in hydroponic and soil systems using the Bengal and Assam Aus Panel. Front Plant Sci 11:576479
Chen F, Fang P, Peng Y, Zeng W, Zhao X, Ding Y et al. (2019) Comparative proteomics of salt-tolerant and salt-sensitive maize inbred lines to reveal the molecular mechanism of salt tolerance. Int J Mol Sci 20:4725
Chen F, Fang P, Zeng W, Ding Y, Zhuang Z, Peng Y (2020) Comparing transcriptome expression profiles to reveal the mechanisms of salt tolerance and exogenous glycine betaine mitigation in maize seedlings. PLoS One 15:p.e0233616
Chen MX, Lu CC, Sun PC, Nie YX, Tian Y, Hu QJ et al. (2021) Comprehensive transcriptome and proteome analyses reveal a novel sodium chloride responsive gene network in maize seed tissues during germination. Plant Cell Environ 44:88–101
Chen R, Cheng Y, Han S, Van Handel B, Dong L, Li X et al. (2017) Whole genome sequencing and comparative transcriptome analysis of a novel seawater adapted, salt-resistant rice cultivar–sea rice 86. BMC Genom 18:1–11
Chen T, Zhu Y, Chen K, Shen C, Zhao X, Shabala S et al. (2020) Identification of new QTL for salt tolerance from rice variety Pokkali. J Agro Crop Sci 206:202–213
Chen T, Shabala S, Niu Y, Chen ZH, Shabala L, Meinke H et al. (2021) Molecular mechanisms of salinity tolerance in rice. Crop J 9:506–520
Cheng X, Liu X, Mao W, Zhang X, Chen S, Zhan K et al. (2018) Genome-wide identification and analysis of HAK/KUP/KT potassium transporters gene family in wheat (Triticum aestivum L.). Int J Mol Sci 19:3969
Choudhary M, Singh A, Rakshit S (2021) Coping with low moisture stress: remembering and responding. Physiol Plant 172:1162–1169
Choudhary M, Wani SH, Kumar P, Bagaria PK, Rakshit S, Roorkiwal M et al. (2019) QTLian breeding for climate resilience in cereals: progress and prospects. Funct Integr Genomics 19:685–701
Church JA, Clark PU, Cazenave A, Gregory JM, Jevrejeva S, Levermann A et al. (2003) Sea level change. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, 1137–1216. https://doi.org/10.1017/CB09781107415315.026
Cui D, Wu D, Somarathna Y, Xu C, Li S, Li P et al. (2015b) QTL mapping for salt tolerance based on SNP markers at the seedling stage in maize (Zea mays L.). Euphytica 203:273–283
Cui D, Wu D, Liu J, Li D, Xu C, Li S et al. (2015a) Proteomic analysis of seedling roots of two maize inbred lines that differ significantly in the salt stress response. PLoS One 10:e0116697
Cui LG, Shan JX, Shi M, Gao JP, Lin HX (2014) The miR156‐SPL 9‐DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J 80:1108–1117
Devi R, Ram S, Rana V, Malik VK, Pande V, Singh GP (2019) QTL mapping for salt tolerance associated traits in wheat (Triticum aestivum L.). Euphytica 215:1–23
Duarte-Delgado D, Dadshani S, Schoof H, Oyiga BC, Schneider M, Mathew B et al. (2020) Transcriptome profiling at osmotic and ionic phases of salt stress response in bread wheat uncovers trait-specific candidate genes. BMC Plant Biol 20:1–18
Dugasa MT, Feng X, Wang NH, Wang J, Wu F (2021) Comparative transcriptome and tolerance mechanism analysis in the two contrasting wheat (Triticum aestivum L.) cultivars in response to drought and salinity stresses. Plant Growth Regul 94:101–14
Egan AN, Schlueter J, Spooner DM (2012) Applications of next‐generation sequencing in plant biology. Am J Bot 99:175–185
El-Hendawy SE, Hassan WM, Al-Suhaibani NA, Refay Y, Abdella KA (2017) Comparative performance of multivariable agro-physiological parameters for detecting salt tolerance of wheat cultivars under simulated saline field growing conditions. Front Plant Sci 8:435
Eren H, Pekmezci M, Okay S, Turktas M, Inal B, Ilhan E et al. (2015) Hexaploid wheat (Triticum aestivum) root miRNome analysis in response to salt stress. Ann Appl Biol 167:208–216
Farooq M, Hussain M, Wakeel A, Siddique KHM (2015) Salt stress in maize: effects, resistance mechanisms, and management. A review. Agron Sustain Dev 35:461–481
Feki K, Quintero FJ, Pardo JM, Masmoudi K (2011) Regulation of durum wheat Na+/H+ exchanger TdSOS1 by phosphorylation. Plant Mol Biol 76:545–56
Ferreira LJ, Azevedo V, Maroco J, Oliveira MM, Santos AP (2015) Salt tolerant and sensitive rice varieties display differential methylome flexibility under salt stress. PLoS One 10:e0124060
Frouin J, Languillaume A, Mas J, Mieulet D, Boisnard A, Labeyrie A et al. (2018) Tolerance to mild salinity stress in japonica rice: a genome-wide association mapping study highlights calcium signaling and metabolism genes. PLoS One 13:e0190964
Fukuda A, Nakamura A, Hara N, Toki S, Tanaka Y (2011) Molecular and functional analyses of rice NHX-type Na+/H+ antiporter genes. Planta 233:175–188
Ganie SA, Molla KA, Henry RJ, Bhat KV, Mondal TK (2019) Advances in understanding salt tolerance in rice. Theor Appl Genet 132:851–870
Garg R, Verma M, Agrawal S, Shankar R, Majee M, Jain M (2014) Deep transcriptome sequencing of wild halophyte rice, Porteresiacoarctata, provides novel insights into the salinity and submergence tolerance factors. DNA Res 21:69–84
Genc Y, Oldach K, Gogel B, Wallwork H, McDonald GK, Smith AB (2013) Quantitative trait loci for agronomic and physiological traits for a bread wheat population grown in environments with a range of salinity levels. Mol Breed 32:39–59
Genc Y, Taylor J, Lyons G, Li Y, Cheong J, Appelbee M et al. (2019) Bread wheat with high salinity and sodicity tolerance. Front Plant Sci 10:1280
Gengmao Z, Shihui L, Xing S, Yizhou W, Zipan C (2015) The role of silicon in physiology of the medicinal plant (Lonicera japonica L.) under salt stress. Sci Rep 5:12696
Van Genuchten MT, Gupta SK (1993) A reassessment of the crop tolerance response function. J Indian Soc Soil Sci 41:730–737
Ghaedrahmati M, Mardi M, Naghavi MR, Majidi Haravan E, Nakhoda B, Azadi A et al. (2014) Mapping QTLs associated with salt tolerance related traits in seedling stage of wheat (Triticum aestivum L.). J Agric Sci Technol 16:1413–1428
Ghomi K, Rabiei B, Sabouri H, Sabouri A (2013) Mapping QTLs for traits related to salinity tolerance at seedling stage of rice (Oryza sativa L.): an agrigenomics study of an Iranian rice population. Omics A J Integrat Biol 17:242–251
Gibbon D (2012) Save and grow: a policymaker’s guide to the sustainable intensification of smallholder crop production. Food and Agriculture Organization of the United Nations, Rome, Italy, p 112. 2011
Gireesh C, Sundaram RM, Anantha SM, Pandey MK, Madhav MS, Rathod S et al. (2021) Nested Association Mapping (NAM) populations: present status and future prospects in the genomics era. Crit Revs Plant Sci 40:49–67
Goddard ME, Hayes BJ (2007) Genomic selection. J Anim breed Genet 124:323–30
Guo R, Yang Z, Li F, Yan C, Zhong X, Liu Q et al. (2015) Comparative metabolic responses and adaptive strategies of wheat (Triticum aestivum) to salt and alkali stress. BMC Plant Biol 15:1–3
Gupta PK, Kulwal PL, Jaiswal V (2014) Association mapping in crop plants: opportunities and challenges. Adv Genet 85:109–148
Gupta PK, Balyan HS, Sharma S, Kumar R (2020) Genetics of yield, abiotic stress tolerance and biofortification in wheat (Triticum aestivum L.). Theor Appl Genet 133:1569–602
Han L, Xiao C, Xiao B, Wang M, Liu J, Bhanbhro N et al. (2019) Proteomic profiling sheds light on alkali tolerance of common wheat (Triticum aestivum L.). Plant Physiol Biochem 138:58–64
Hanin M, Ebel C, Ngom M, Laplaze L, Masmoudi K (2016) New insights on plant salt tolerance mechanisms and their potential use for breeding. Front Plant Sci 7:1787
He Y, Li W, Lv J, Jia Y, Wang M, Xia G (2012) Ectopic expression of a wheat MYB transcription factor gene, TaMYB73, improves salinity stress tolerance in Arabidopsis thaliana. J Exp Bot 63:1511–1522
Hernández JA (2019) Salinity tolerance in plants: trends and perspectives. Int J Mol Sci 20:2408
Ho VT, Thomson MJ, Ismail AM (2016) Development of salt tolerant IR64 near isogenic lines through marker-assisted breeding. J Crop Sci Biotechnol 19:373–381
Hoque ABMZ, Haque MA, Sarker MRA, Rahman MA (2015) Marker-assisted introgression of Saltol locus into genetic background of BRRI Dhan-49. Int J Biosci 6:71–80
Hossain H, Rahman MA, Alam MS, Singh RK (2015) Mapping of quantitative trait loci associated with reproductive‐stage salt tolerance in rice. J Agron Crop Sci 201:17–31
Hu P, Zheng Q, Luo Q, Teng W, Li H, Li B et al. (2021) Genome-wide association study of yield and related traits in common wheat under salt-stress conditions. BMC Plant Biol 21:27
Hussain B, Lucas SJ, Ozturk L, Budak H (2017) Mapping QTLs conferring salt tolerance and micronutrient concentrations at seedling stage in wheat. Sci Rep 7:1–4
Hussein MM, Balbaa LK, Gaballah MS (2007) Salicylic acid and salinity effects on growth of maize plants. Res J Agric Biol Sci 3:321–328
Huyen LTN, Cuc LM, Ismail AM, Ham LH (2012) Introgression the salinity tolerance QTLs saltol into AS996, the elite rice variety of Vietnam. Am J Plant Sci 3:981–987
Huyen LTN, Cuc LM, Ham LH, Khanh TD (2013) Introgression the saltol QTL into Q5DB, the elite variety of Vietnam using marker-assisted-selection (MAS). Am J Bio Sci 1:80–84
Ilyas N, Amjid MW, Saleem MA, Khan W, Wattoo FM, Rana RM et al. (2020) Quantitative trait loci (QTL) mapping for physiological and biochemical attributes in a Pasban90/Frontana recombinant inbred lines (RILs) population of wheat (Triticum aestivum) under salt stress condition. Saudi J Biol Sci 27:341–51
Iqbal S, Hussain S, Qayyaum MA, Ashraf M (2020) The response of maize physiology under salinity stress and its coping strategies. In: Plant Stress Physiology, Intech Open https://doi.org/10.5772/intechopen.92213
Islam M, Ontoy J, Subudhi PK (2019) Meta-analysis of quantitative trait loci associated with seedling-stage salt tolerance in rice (Oryza sativa L.). Plants 8:33
Jacoby RP, Millar AH, Taylor NL (2013) Investigating the role of respiration in plant salinity tolerance by analyzing mitochondrial proteomes from wheat and a salinity-tolerant Amphiploid (wheat × Lophopyrumelongatum). J Proteome Res 12:4807–4829
Jahan N, Zhang Y, Lv Y, Song M, Zhao C, Hu H et al. (2020) QTL analysis for rice salinity tolerance and fine mapping of a candidate locus qSL7 for shoot length under salt stress. Plant Growth Regul 90:307–319
Jahani M, Mohammadi-Nejad G, Nakhoda B, Rieseberg LH (2019) Genetic dissection of epistatic and QTL by environment interaction effects in three bread wheat genetic backgrounds for yield-related traits under saline conditions. Euphytica 215:103
James RA, Blake C, Byrt CS, Munns R (2011) Major genes for Na+ exclusion, Nax1 and Nax2 (wheat HKT1; 4 and HKT1; 5), decrease Na+ accumulation in bread wheat leaves under saline and waterlogged conditions. J Exp Bot 62:2939–2947
Jha UC, Bohra A, Jha R, Parida SK (2019) Salinity stress response and ‘omics’ approaches for improving salinity stress tolerance in major grain legumes. Plant Cell Rep 38:255–277
Jiang C, Zheng Q, Liu Z, Xu W, Liu L, Zhao G, Long X (2012) Overexpression of Arabidopsis thaliana Na+/H+ antiporter gene enhanced salt resistance in transgenic poplar (Populus × euramericana “Neva”). Trees 26:685–694
Jiang Q, Hu Z, Zhang H, Ma Y (2014) Overexpression of GmDREB1 improves salt tolerance in transgenic wheat and leaf protein response to high salinity. Crop J 2:120–131
Jiang Q, Li X, Niu F, Sun X, Hu Z, Zhang H (2017) iTRAQ‐based quantitative proteomic analysis of wheat roots in response to salt stress. Proteomics 17:1600265
Jiang Z, Song G, Shan X, Wei Z, Liu Y, Jiang C et al. (2018) Association analysis and identification of ZMHKT1;5 variation with salt-stress tolerance. Front Plant Sci 9:1485
Kamal AHM, Cho K, Kim DE, Uozumi N, Chung KY, Lee SY et al. (2012) Changes in physiology and protein abundance in salt-stressed wheat chloroplasts. Mol Biol Rep 39:9059–9074
Karan R, DeLeon T, Biradar H, Subudhi PK (2012) Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS One 7:e40203
Kashyap M, Ford R, Bohra A, Kuvalekar A, Mantri N (2017) Improving salt tolerance of chickpea using modern genomics tools and molecular breeding. Curr Genom 18:557–567
Kashyap SP, Prasanna HC, Kumari N, Mishra P, Singh B (2020) Understanding salt tolerance mechanism using transcriptome profiling and de novo assembly of wild tomato Solanum chilense. Sci rep 10:1–20
Kaur S, Iquebal MA, Jaiswal S, Tandon G, Sundaram RM, Gautam RK et al. (2016) A meta-analysis of potential candidate genes associated with salinity stress tolerance in rice. Agric Gene 1:126–134
Khan AW, Garg V, Roorkiwal M, Golicz AA, Edwards D, Varshney RK (2020) Super-pangenome by integrating the wild side of a species for accelerated crop improvement. Trends Plant Sci 25:148–58
Khan MSK, Saeed M, Iqbal J (2015) Identification of quantitative trait loci for Na+, K+ and Ca++ accumulation traits in rice grown under saline conditions using F2 mapping population. Braz J Bot 38:555–565
Khan MSK, Saeed M, Iqbal J (2016) Quantitative trait locus mapping for salt tolerance at maturity stage in indica rice using replicated F2 population. Braz J Bot 39:641–650
Kong W, Zhong H, Gong Z, Fang X, Sun T, Deng X et al. (2019) Meta-analysis of salt stress transcriptome responses in different rice genotypes at the seedling stage. Plants 8:64
Kotula L, Garcia Caparros P, Zörb C, Colmer TD, Flowers TJ (2020) Improving crop salt tolerance using transgenic approaches: an update and physiological analysis. Plant Cell Environ 43(12):2932–2956
Kravchik M, Bernstein N (2013) Effects of salinity on the transcriptome of growing maize leaf cells point at cell-age specificity in the involvement of the antioxidative response in cell growth restriction. BMC Genom 14:1–13
Krishnamurthy SL, Pundir P, Warraich AS, Rathor S, Lokeshkumar BM, Singh NK et al. (2020) Introgressed saltol QTL lines improves the salinity tolerance in rice at seedling stage. Front Plant Sci 11:833
Kumar K, Gambhir G, Dass A, Tripathi AK, Singh A, Jha AK et al. (2020) Genetically modified crops: current status and future prospects. Planta 251:1–27
Kumar P, Sharma PK (2020) Soil salinity and food security in India. Front Sustain Food Syst 4:533781
Kumar S, Beena AS, Awana M, Singh A (2017) Physiological, biochemical, epigenetic and molecular analyses of wheat (Triticum aestivum) genotypes with contrasting salt tolerance. Front Plant Sci 8:1151
Kumar V, Singh A, Mithra SA, Krishnamurthy SL, Parida SK, Jain S et al. (2015) Genome-wide association mapping of salinity tolerance in rice (Oryza sativa). DNA Res 22:133–145
Kumar VS, Verma RK, Yadav SK, Yadav P, Watts A, Rao MV et al. (2020) CRISPR-Cas9 mediated genome editing of drought and salt tolerance (OsDST) gene in indica mega rice cultivar MTU1010. Physiol Mol Biol Plants 26:1099
Kumari S, Nee Sabharwal VP, Kushwaha HR, Sopory SK, Singla-Pareek SL, Pareek A (2009) Transcriptome map for seedling stage specific salinity stress response indicates a specific set of genes as candidate for saline tolerance in Oryza sativa L. Funct Integr Genom 9:109–123
Kurotani KI, Hayashi K, Hatanaka S, Toda Y, Ogawa D, Ichikawa H et al. (2015) Elevated levels of CYP94 family gene expression alleviate the jasmonate response and enhance salt tolerance in rice. Plant Cell Physiol 56:779–89
Le TD, Gathignol F, Vu HT, Nguyen KL, Tran LH, Vu HTT et al. (2021) Genome-wide association mapping of salinity tolerance at the seedling stage in a panel of Vietnamese landraces reveals new valuable QTLs for salinity stress tolerance breeding in rice. Plants 10:1088
Lei L, Zheng H, Bi Y, Yang L, Liu H, Wang J et al. (2020) Identification of a major QTL and candidate gene analysis of salt tolerance at the bud burst stage in rice (Oryza sativa L.) using QTL-Seq and RNA-Seq. Rice 13:1–14
Lekklar C, Pongpanich M, Suriya-arunroj D, Chinpongpanich A, Tsai H, Comai L et al. (2019) Genome-wide association study for salinity tolerance at the flowering stage in a panel of rice accessions from Thailand. BMC Genom 20:76
De Leon TB, Linscombe S, Subudhi PK (2016) Molecular dissection of seedling salinity tolerance in rice (Oryza sativa L.) using a high-density GBS-based SNP linkage map. Rice 9:1–22
De Leon TB, Linscombe S, Subudhi PK (2017) Identification and validation of QTLs for seedling salinity tolerance in introgression lines of a salt tolerant rice landrace ‘Pokkali’. PLoS One 12:e0175361
Li P, Cao W, Fang H, Xu S, Yin S, Zhang Y et al. (2017) Transcriptomic profiling of the maize (Zea mays L.) leaf response to abiotic stresses at the seedling stage. Front Plant Sci 8:290
Li Q, Ma C, Tai H, Qiu H, Yang A (2020) Comparative transcriptome analysis of two rice genotypes differing in their tolerance to saline-alkaline stress. PLoS One 15:e0243112
Li YF, Zheng Y, Vemireddy LR, Panda SK, Jose S, Ranjan A et al. (2018) Comparative transcriptome and translatome analysis in contrasting rice genotypes reveals differential mRNA translation in salt-tolerant Pokkali under salt stress. BMC Genom 19:95–113
Liang X, Liu S, Wang T, Li F, Cheng J, Lai J et al. (2021) Metabolomics-driven gene mining and genetic improvement of tolerance to salt-induced osmotic stress in maize. N Phytol 230:2355–2370
Linh TH, Xuan TD, Ham LH, Ismail AM, Khanh TD (2012) Molecular breeding to improve salt tolerance of rice (Oryza sativa L.) in the Red River Delta of Vietnam. Int J plant Genom 2012:949038
Liu C, Li S, Wang M, Xia G (2012) A transcriptomic analysis reveals the nature of salinity tolerance of a wheat introgression line. Plant Mol Biol 78:159–169
Liu C, Chen K, Zhao X, Wang X, Shen C, Zhu Y et al. (2019) Identification of genes for salt tolerance and yield-related traits in rice plants grown hydroponically and under saline field conditions by genome-wide association study. Rice 12:1–13
Lu W, Guo C, Li X, Duan W, Ma C, Zhao M et al. (2014) Overexpression of TaNHX3, a vacuolar Na+/H+ antiporter gene in wheat, enhances salt stress tolerance in tobacco by improving related physiological processes. Plant Physiol Biochem 76:17–28
Luo M, Zhao Y, Zhang R, Xing J, Duan M, Li J et al. (2017) Mapping of a major QTL for salt tolerance of mature field-grown maize plants based on SNP markers. BMC Plant Biol 17:140
Luo M, Zhao Y, Wang Y, Shi Z, Zhang P, Zhang Y et al. (2018) Comparative proteomics of contrasting maize genotypes provides insights into salt-stress tolerance mechanisms. J Proteome Res 17:141–153
Luo M, Zhang Y, Chen K, Kong M, Song W, Lu B et al. (2019) Mapping of quantitative trait loci for seedling salt tolerance in maize. Mol Breed 39:1–12
Luo M, Zhang Y, Li J, Zhang P, Chen K, Song W et al. (2021) Molecular dissection of maize seedling salt tolerance using a genome-wide association analysis method. Plant Biotechnol J https://doi.org/10.1111/pbi.13607
Lv DW, Zhu GR, Zhu D, Bian YW, Liang XN, Cheng ZW et al. (2016) Proteomic and phosphoproteomic analysis reveals the response and defense mechanism in leaves of diploid wheat T. monococcum under salt stress and recovery. J Proteom 143:93–105
Ma Q, Shi C, Su C, Liu Y (2020) Complementary analyses of the transcriptome and iTRAQ proteome revealed mechanism of ethylene dependent salt response in bread wheat (Triticum aestivum L.). Food Chem 325:126866
Ma Q, Zhou H, Sui X, Su C, Yu Y, Yang H et al. (2021) Generation of new salt-tolerant wheat lines and transcriptomic exploration of the responsive genes to ethylene and salt stress. Plant Growth Regul 94:33–48
Mahajan MM, Goyal E, Singh AK, Gaikwad K, Kanika K (2020) Shedding light on response of Triticum aestivum cv. Kharchia Local roots to long-term salinity stress through transcriptome profiling. Plant Growth Regul 90:369–381
Maleki M, Naghavi MR, Alizadeh H, Poostini K, Abd Mishani C (2014) Comparison of protein changes in the leaves of two bread wheat cultivars with different sensitivity under salt stress. Ann Res Rev Biol 15:1784–97
Manohara KK, Morajkar S, Shanbhag Y, Phadte P, Singh NK (2021) Haplotype analysis of Saltol QTL region in diverse landraces, wild rice and introgression lines of rice (Oryza sativa L.). Plant Genet Resour 19: 289–98
Mansuri RM, Shobbar ZS, Jelodar NB, Ghaffari M, Mohammadi SM, Daryani P (2020) Salt tolerance involved candidate genes in rice: an integrative meta-analysis approach. BMC Plant Biol 20:1–14
Martínez-Atienza J, Jiang X, Garciadeblas B, Mendoza I, Zhu JK, Pardo JM et al. (2007) Conservation of the salt overly sensitive pathway in rice. Plant Physiol 143:1001–1012
Masoudi B, Mardi M, Hervan EM, Bihamta MR, Naghavi MR, Nakhoda B et al. (2015) QTL mapping of salt tolerance traits with different effects at the seedling stage of bread wheat. Plant Mol Biol Rep 33:1790–803
Mazumder A, Rohilla M, Bisht DS, Krishnamurthy SL, Barman M, Sarma RN et al. (2020) Identification and mapping of quantitative trait loci (QTL) and epistatic QTL for salinity tolerance at seedling stage in traditional aromatic short grain rice landrace Kolajoha (Oryza sativa L.) of Assam, India. Euphytica 216:1–18
Meena KK, Bitla UM, Sorty AM, Singh DP, Gupta VK, Wakchaure GC et al. (2020) Mitigation of salinity stress in wheat seedlings due to the application of phytohormone-rich culture filtrate extract of methylotrophic actinobacterium Nocardioides sp. NIMMe6. Front Microbiol 11, https://doi.org/10.3389/fmicb.2020.02091
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA 88:9828–9832
Mittal S, Kumari N, Sharma V (2012) Differential response of salt stress on Brassica juncea: photosynthetic performance, pigment, proline, D1 and antioxidant enzymes. Plant Physiol Biochem 54:17–26
Mohammadi R, Mendioro MS, Diaz GQ, Gregorio GB, Singh RK (2013) Mapping quantitative trait loci associated with yield and yield components under reproductive stage salinity stress in rice (Oryza sativa L.). J Genet 92:433–443
Moniruzzaman M, Islam MS, Rashid JA, Begum SN, Islam MM (2012) Marker-assisted backcrossing for identification of salt tolerant rice lines. Int J Agric Res Innov Technol 2:1–8
Moradi F, Ismail AM, Egdane J, Gregorio GB (2003) Salinity tolerance of rice during reproductive development and association with tolerance at the seedling stage. Ind J Plant Physiol 8:105–116
Munns R (2002) Comparative physiology of salt and water stress. Plant Cell Environ 25:239–250
Munns R, Tester M (2008) Mechanisms of salinity tolerance. Ann Rev Plant Biol 59:651–681
Munns R, James R, Xu B, Athman A, Conn SJ, Jordans C et al. (2012) Wheat grain yield on saline soils is improved by an ancestral Na+ transporter gene. Nat Biotechnol 30:360–364
Nabati J, Kafi M, Nezami A, Moghaddam PR, Ali M, Mehrjerdi MZ (2011) Effect of salinity on biomass production and activities of some key enzymatic antioxidants in Kochia (Kochia scoparia). Pak J Bot 43:539–548
Nair MM, Shylaraj KS (2021) Introgression of dual abiotic stress tolerance QTLs (Saltol QTL and Sub1 gene) into Rice (Oryza sativa L.) variety Aiswarya through marker-assisted backcross breeding. Physiol Mol Biol Plants 27:497–514
Nam MH, Bang E, Kwon TY, Kim Y, Kim EH, Cho K et al. (2015) Metabolite profiling of diverse rice germplasm and identification of conserved metabolic markers of rice roots in response to long-term mild salinity stress. Int J Mol Sci 16:21959–21974
Narjesi V, Mardi M, Hervan EM, Azadi A, Naghavi MR, Ebrahimi M et al. (2015) Analysis of quantitative trait loci (QTL) for grain yield and agronomic traits in wheat (Triticum aestivum L.) under normal and salt-stress conditions. Plant Mol Biol Rep 33:2030–40
Naveed SA, Zhang F, Zhang J, Zheng TQ, Meng LJ, Pang YL et al. (2018) Identification of QTLs and candidate genes for salinity tolerance at the germination and seedling stages in rice by genome-wide association analyses. Sci Rep 8:6505
Nayyeripasand L, Garoosi GA, Ahmadikhah A (2021) Genome-wide association study (GWAS) to identify salt-tolerance QTLs carrying novel candidate genes in rice during early vegetative stage. Rice 14:1–21
Nicholls RJ, Marinova N, Lowe JA, Brown S, Vellinga P, De Gusmão D et al. (2011) Sea-level rise and its possible impacts given a ‘beyond 4C world’ in the twenty first century. Philos Trans R Soc A Math Phys Eng Sci 369:161–181
Oyiga BC, Sharma RC, Shen J, Baum M, Ogbonnaya FC, Léon J et al. (2016) Identification and characterization of salt tolerance of wheat germplasm using a multivariable screening approach. J Agron Crop Sci 202:472–485
Oyiga BC, Sharma RC, Baum M, Ogbonnaya FC, Léon J, Ballvora A (2018) Allelic variations and differential expressions detected at quantitative trait loci for salt stress tolerance in wheat. Plant Cell Environ 41:919–935
Oyiga BC, Ogbonnaya FC, Sharma RC, Baum M, Léon J, Ballvora A (2019) Genetic and transcriptional variations in NRAMP-2 and OPAQUE1 genes are associated with salt stress response in wheat. Theor Appl Genet 132:323–346
Pandit A, Rai V, Bal S, Sinha S, Kumar V, Chauhan M et al. (2010) Combining QTL mapping and transcriptome profiling of bulked RILs for identification of functional polymorphism for salt tolerance genes in rice (Oryza sativa L.). Mol Genet Genom 284:121–136
Parvaiz A, Latef A, Saiema R, Akram NA, Muhammad A, Salih G (2016) Role of proteomics in crop stress tolerance. Front Plant Sci 7: https://doi.org/10.3389/fpls.2016.01336
Prakash NR, Sheoran S, Saini M, Punia M, Rathod NKK, Bhinda MS et al. (2020). Offsetting climate change impact through genetic enhancement. In: Srinivasrao C, et al. (ed) Climate Change and Indian Agriculture: Challenges and Adaptation strategies. ICAR-National Academy of Agricultural Research Management, Hyderabad, Telangana, India, pp 71–104
Pundir P, Devi A, Krishnamurthy SL, Sharma PC, Vinay Kumar NM (2021) QTLs in salt rice variety CSR10 reveals salinity tolerance at reproductive stage. Physiol Plant 43:1–15
Punyawaew K, Suriya-Arunroj D, Siangliw M, Thida M, Lanceras-Siangliw J, Fukai S et al. (2016) Thai jasmine rice cultivar KDML105 carrying Saltol QTL exhibiting salinity tolerance at seedling stage. Mol Breed 36:1–13
Puram VRR, Ontoy J, Subudhi PK (2018) Identification of QTLs for salt tolerance traits and pre breeding lines with enhanced salt tolerance in an introgression line population of rice. Plant Mol Biol Rep. 36:695–709
Puram VRR, Ontoy J, Linscombe S, Subudhi PK (2017) Genetic dissection of seedling stage salinity tolerance in rice using introgression lines of a salt tolerant landrace Nona Bokra. J Heredity 108:658–670
Qiu X, Yuan Z, Liu H, Xiang X, Yang L, He W et al. (2015) Identification of salt tolerance‐improving quantitative trait loci alleles from a salt‐susceptible rice breeding line by introgression breeding. Plant Breed 134:653–660
Quilis J, Peñas G, Messeguer J, Brugidou C, Segundo BS (2008) The Arabidopsis AtNPR1 inversely modulates defense responses against fungal, bacterial, or viral pathogens while conferring hypersensitivity to abiotic stresses in transgenic rice. Mol Plant Microbe Interact 21:1215–1231
Quintero JM, Fournier JM, Benlloch M (2007) Na+ accumulation in shoot is related to water transport in K+-starved sunflower plants but not in plants with a normal K+ status. J Plant Physiol 164:60–67
Rahman MA, Bimpong IK, Bizimana JB, Pascual ED, Arceta M, Swamy BM et al. (2017) Mapping QTLs using a novel source of salinity tolerance from Hasawi and their interaction with environments in rice. Rice 10:1–17
Rahman MA, Thomson MJ, De Ocampo M, Egdane JA, Salam MA, Shah-E-Alam M et al. (2019) Assessing trait contribution and mapping novel QTL for salinity tolerance using the Bangladeshi rice landrace Capsule. Rice 12:1–18
Rajkumar MS, Shankar R, Garg R, Jain M (2020) Bisulphite sequencing reveals dynamic DNA methylation under desiccation and salinity stresses in rice cultivars. Genomics 112:3537–3548
Rana MM, Takamatsu T, Baslam M, Kaneko K, Itoh K, Harada N et al. (2019) Salt tolerance improvement in rice through efficient SNP marker-assisted selection coupled with speed-breeding. Int J Mol Sci 20:2585
Ren ZH, Gao JP, Li LG, Cai XL, Huang W, Chao DY et al. (2005) A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat Genet 37:1141–1146
Ren Y, Xu Y, Teng W, Li B, Lin T (2018) QTLs for seedling traits under salinity stress in hexaploid wheat. Ciencia Rural 48: https://doi.org/10.1590/0103-8478cr20170446
Rhoades JD, Kandiah A, Mashali AM (1992) The use of saline waters for crop production. FAO Irrigation and Drainage Paper, Food and Agriculture Organization of the United Nations, Rome, Italy, ISBN 978-92-5-103237-4
Sandhu D, Pudussery MV, Kumar R, Pallete A, Markley P, Bridges WC et al. (2020) Characterization of natural genetic variation identifies multiple genes involved in salt tolerance in maize. Funct Integr Genom https://doi.org/10.1007/s10142-019-00707-x
Sapre S, Gontia-Mishra I, Tiwari S (2018) Klebsiella sp. confers enhanced tolerance to salinity and plant growth promotion in oat seedlings (Avena sativa). Microbiol Res 206:25–32
Shahid MA, Sarkhosh A, Khan N, Balal RM, Ali S, Rossi L et al. (2020) Insights into the physiological and biochemical impacts of salt stress on plant growth and development. Agronomy 10:938
Shahverdi MA, Omidi H, Tabatabaei SJ (2018) Plant growth and steviol glycosides as affected by foliar application of selenium, boron, and iron under NaCl stress in Stevia rebaudiana Bertoni. Ind Crops Prod 125:408–415
Shakirova FM, Sakhabutdinova AR, Bezrukova MV, Fatkhutdinova RA, Fatkhutdinova DR (2003) Changes in the hormonal status of wheat seedlings induced by salicylic acid and salinity. Plant Sci 164:317–22
Shamaya NJ, Shavrukov Y, Langridge P, Roy SJ, Tester M (2017) Genetics of Na+ exclusion and salinity tolerance in Afghani durum wheat landraces. BMC Plant Biol 17:1–8
Shankar R, Bhattacharjee A, Jain M (2016) Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci Rep 6:1–15
Sharma H, Taneja M, Upadhyay SK (2020) Identification, characterization and expression profiling of cation-proton antiporter superfamily in Triticum aestivum L. and functional analysis of TaNHX4-B. Genomics 112:356–370
Shrivastava P, Kumar R (2015) Soil salinity: a serious environmental issue and plant growth promoting bacteria as one of the tools for its alleviation. Saudi J Biol Sci 22:123–131
Singh K, Batra R, Sharma S, Saripalli G, Gautam T, Singh R et al. (2021) WheatQTLdb: a QTL database for wheat. Mol Genet Genom 296(5):1051–1056. https://doi.org/10.1007/s00438-021-01796-9
Singh RK, Gregorio GB, Jain RK (2007) QTL mapping for salinity tolerance in rice. Physiol Mol Biol Plants 13:87
Singh RP, Runthala A, Khan S, Jha PN (2017) Quantitative proteomics analysis reveals the tolerance of wheat to salt stress in response to Enterobacter cloacae SBP-8. PLoS One 12:e0183513
Singh RK, Kota S, Flowers TJ (2021) Salt tolerance in rice: seedling and reproductive stage QTL mapping come of age. Theor Appl Genet. https://doi.org/10.1007/s00122-021-03890-3
Singh VK, Singh BD, Kumar A, Maurya S, Krishnan SG, Vinod KK et al. (2018) Marker-assisted introgression of Saltol QTL enhances seedling stage salt tolerance in the rice variety “Pusa Basmati 1”. Int J Genom https://doi.org/10.1155/2018/8319879
Sinha P, Singh VK, Saxena RK, Khan AW, Abbai R, Chitikineni A et al. (2020) Superior haplotypes for haplotype based breeding for drought tolerance in pigeonpea (Cajanus cajan L.). Plant Biotechnol J 18:2482–2490
Sinha P, Singh VK, Bohra A, Kumar A, Reif JC, Varshney RK (2021) Genomics and breeding innovations for enhancing genetic gain for climate resilience and nutrition traits. Theor Appl Genet 134:1829–1843
Soares AL, Geilfus CM, Carpentier SC (2018) Genotype-specific growth and proteomic responses of maize toward salt stress. Front Plant Sci 9:661
Sun BR, Fu CY, Fan ZL, Chen Y, Chen WF, Zhang J et al. (2019) Genomic and transcriptomic analysis reveal molecular basis of salinity tolerance in a novel strong salt-tolerant rice landrace Changmaogu. Rice 12:1–15
Sun L, Miao X, Cui J, Deng J, Wang X, Wang Y et al. (2018) Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across different salt stress in Maize (Zea mays L.) Euphytica 214:1–5
Suryendra PJ, Revathi P, Singh AK, Viraktamath BC (2020) Marker-assisted recurrent selection for genetic male sterile population improvement in rice. Electron J Plant Breed 11:149–155
Takahashi F, Tilbrook J, Trittermann C, Berger B, Roy SJ, Seki M et al. (2015) Comparison of leaf sheath transcriptome profiles with physiological traits of bread wheat cultivars under salinity stress. PLoS One 10:e0133322
Thanasilungura K, Kranto S, Monkham T, Chankaew S, Sanitchon J (2020) Improvement of a RD6 rice variety for blast resistance and salt tolerance through marker-assisted backcrossing. Agronomy 10:1118
Tiwari S, Krishnamurthy SL, Kumar V, Singh B, Rao AR, Mithra SVA et al. (2016) Mapping QTLs for salt tolerance in rice (Oryza sativa L.) by bulked segregant analysis of recombinant inbred lines using 50K SNP chip. PLoS One 11:e0153610
Tnay G (2019) Too much salt: the growing threat that salinity poses to global food production. Future Directions International Pty Ltd, Dalkeith
Valarmathi M, Sasikala R, Rahman H, Jagadeeshselvam N, Kambale R, Raveendran M (2019) Development of salinity tolerant version of a popular rice variety improved white ponni through marker assisted back cross breeding. Plant Physiol Rep 24:262–271
Wang M, Wang Y, Zhang Y, Li C, Gong S, Yan S et al. (2019) Comparative transcriptome analysis of salt-sensitive and salt-tolerant maize reveals potential mechanisms to enhance salt resistance. Genes Genom 41:781–801
Wang W, Vinocur B, Altman A (2003) Plant responses to drought, salinity and extreme temperatures: towards genetic engineering for stress tolerance. Planta 218:1–14
Wang WS, Pan YJ, Zhao XQ, Dwivedi D, Zhu LH, Ali J et al. (2011) Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J Exp Bot 62:1951–1960
Wang Y, Huang L, Du F, Wang J, Zhao X, Li Z et al. (2021) Comparative transcriptome and metabolome profiling reveal molecular mechanisms underlying OsDRAP1-mediated salt tolerance in rice. Sci Rep 11:1–11
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang Z, Cheng J, Chen Z, Huang J, Bao Y, Wang J et al. (2012) Identification of QTLs with main, epistatic and QTL × environment interaction effects for salt tolerance in rice seedlings under different salinity conditions. Theor Appl Genet 125:807–815
Warraich AS, Krishnamurthy SL, Sooch BS, Vinaykumar NM, Dushyanth kumar BM, Bose J et al. (2020) Rice GWAS reveals key genomic regions essential for salinity tolerance at reproductive stage. Acta Physiol Plant 42(8):1–15
Watson A, Ghosh S, Williams MJ, William SC, Simmonds J, Rey MD et al. (2018) Speed breeding is a powerful tool to accelerate crop research and breeding. Nat Plants 4:23–29
Wu F, Yang J, Yu D, Xu P (2020) Identification and validation a major QTL from “Sea Rice 86” seedlings conferred salt tolerance. Agronomy 10:410
Xie Z, Wang J, Wang W, Wang Y, Xu J, Li Z et al. (2021) Integrated analysis of the transcriptome and metabolome revealed the molecular mechanisms underlying the enhanced salt tolerance of rice due to the application of exogenous melatonin. Front Plant Sci 11:618–680
Xu H, Jiang X, Zhan K, Cheng X, Chen X, Pardo JM et al. (2008) Functional characterization of a wheat plasma membrane Na+/H+ antiporter in yeast. Arch Biochem Biophys 473:8–15
Xu Y, Zhou Y, Hong S, Xia Z, Cui D, Guo J et al. (2013) Functional characterization of a wheat NHX antiporter gene TaNHX2 that encodes a K+/H+ exchanger. PLoS One 8:e78098
Xu YF, An DG, Liu DC, Zhang AM, Xu HX, Li B (2012) Mapping QTLs with epistatic effects and QTL × treatment interactions for salt tolerance at seedling stage of wheat. Euphytica 186:233–245
Yadav AK, Kumar A, Grover N, Ellur RK, Krishnan SG, Bollinedi H et al. (2020) Marker aided introgression of ‘Saltol’, a major QTL for seedling stage salinity tolerance into an elite Basmati rice variety ‘Pusa Basmati 1509’. Sci Rep 10:1–15
Yan M, Zheng L, Li B, Shen R, Lan P (2020) Comparative proteomics reveals new insights into the endosperm responses to drought, salinity and submergence in germinating wheat seeds. Plant Mol Biol 105(3):287–302. https://doi.org/10.1007/s11103-020-01087-8
Yang T, Zhang S, Hu Y, Wu F, Hu Q, Chen G et al. (2014) The role of a potassium transporter OsHAK5 in potassium acquisition and transport from roots to shoots in rice at low potassium supply levels. Plant Physiol 166:945–959
Yang T, Feng H, Zhang S, Xiao H, Hu Q, Chen G et al. (2020) The potassium transporter OsHAK5 alters rice architecture via ATP-dependent transmembrane auxin fluxes. Plant Commun 1:100052
Yang Y, Guo Y (2018) Unraveling salt stress signaling in plants. J Integr Plant Biol 60:796–804
Yuan J, Wang X, Zhao Y, Khan NU, Zhao Z, Zhang Y et al. (2020) Genetic basis and identification of candidate genes for salt tolerance in rice by GWAS. Sci Rep 10:9958
Yue JY, Wang LH, Dou XT, Wang YJ, Wang HZ (2020) Comparative metabolomic profiling in the roots of salt-tolerant and salt-intolerant maize cultivars treated with NaCl stress. Biol Plant 64:569–577
Van Zelm E, Zhang Y, Testerink C (2020) Salt tolerance mechanisms of plants. Annu Rev Plant Biol 71:403–433
Zhang A, Liu Y, Wang F, Li T, Chen Z, Kong D et al. (2019) Enhanced rice salinity tolerance via CRISPR/Cas9-targeted mutagenesis of the OsRR22 gene. Mol Breed 39:1–10
Zhang M, Cao Y, Wang Z, Wang ZQ, Shi J, Liang X et al. (2018) A retrotransposon in an HKT1 family sodium transporter causes variation of leaf Na+ exclusion and salt tolerance in maize. N Phytol 217:1161–1176
Zhang X, Liu P, Qing C, Yang C, Shen Y, Ma L (2021) Comparative transcriptome analyses of maize seedling root responses to salt stress. Peer J 9:e10765
Zhang Y, Feng D, Bao Y, Xin M, Yin N, Xu J et al. (2013) A novel wheat related-to-Ubiquitin gene TaRUB1 is responsive to pathogen attack as well as to both osmotic and salt stress. Plant Mol Biol Rep 31:151–159
Zhang Z, Zhang J, Chen Y, Li R, Wang H, Wei J (2012) Genome-wide analysis and identification of HAK potassium transporter gene family in maize (Zea mays L.). Mol Biol Rep 39:8465–8473
Zheng H, Zhao H, Liu H, Wang J, Zou D (2015) QTL analysis of Na+ and K+ concentrations in shoots and roots under NaCl stress based on linkage and association analysis in japonica rice. Euphytica 201:109–121
Zhou Y, Yang P, Cui F, Zhang F, Luo X, Xie J (2016) Transcriptome analysis of salt stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). PLoS One 11:e0146242
Zhu D, Luo F, Zou R, Liu J, Yan Y (2021) Integrated physiological and chloroplast proteome analysis of wheat seedling leaves under salt and osmotic stresses. J Proteom 234:104097
Zhu JK (2001) Plant salt tolerance. Trends Plant Sci 6:66–71
Zörb C, Noll A, Karl S, Leib K, Yan F, Schubert S (2005) Molecular characterization of Na+/H+ antiporters (ZmNHX) of maize (Zea mays L.) and their expression under salt stress. J Plant Physiol 162:55–66
Zörb C, Schmitt S, Mühling KH (2010) Proteomic changes in maize roots after short-term adjustment to saline growth conditions. Proteomics 10:4441–4444
Acknowledgements
The authors thank the Indian Council of Agricultural Research (ICAR) for support received in preparing the manuscript. The first author thanks the Science and Engineering Research Board (SERB) Government of India for financial support through the EEQ (2018/001394) scheme.
Author information
Authors and Affiliations
Contributions
PK and MC conceptualized the manuscript. PK, MC, TH, NRP, VS, VTV, SS, NL and RKT interpreted the literature and drafted the manuscript: section wise. SR and KHMS critically edited the manuscript. PK, MC and NRP contributed equally in the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Kumar, P., Choudhary, M., Halder, T. et al. Salinity stress tolerance and omics approaches: revisiting the progress and achievements in major cereal crops. Heredity 128, 497–518 (2022). https://doi.org/10.1038/s41437-022-00516-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00516-2
This article is cited by
-
Unveiling the dynamic relationship of viruses and/or symbiotic bacteria with plant resilience in abiotic stress
Stress Biology (2024)
-
Integrated single-molecule real-time sequencing and RNA sequencing reveal the molecular mechanisms of salt tolerance in a novel synthesized polyploid genetic bridge between maize and its wild relatives
BMC Genomics (2023)
-
Characterization of ZmPMP3g function in drought tolerance of maize
Scientific Reports (2023)
-
An integrated system with functions of solar desalination, power generation and crop irrigation
Nature Water (2023)
-
Signal crosstalk of phytomelatonin during salinity stress tolerance in plants
Plant Growth Regulation (2023)
