
Abstract
Deformities in cultured fish species may be genetic, and identifying causative genes is essential to expand production and maintain farmed animal welfare. We previously reported a genetic deformity in juvenile red sea bream, designated a transparent phenotype. To identify its causative gene, we conducted genome-wide linkage analysis and identified two single nucleotide polymorphisms (SNP) located on LG23 directly linked to the transparent phenotype. The scaffold on which the two SNPs were located contained two candidate genes, duox and duoxa, which are related to thyroid hormone synthesis. Four missense mutations were found in duox and one in duoxa, with that in duoxa showing perfect association with the transparent phenotype. The mutation of duoxa was suggested to affect the transmembrane structure and thyroid-related traits, including an enlarged thyroid gland and immature erythrocytes, and lower thyroxine (T4) concentrations were observed in the transparent phenotype. The transparent phenotype was rescued by T4 immersion. Loss-of-function of duoxa by CRISPR–Cas9 induced the transparent phenotype in zebrafish. Evidence suggests that the transparent phenotype of juvenile red sea bream is caused by the missense mutation of duoxa and that this mutation disrupts thyroid hormone synthesis. The newly identified missense mutation will contribute to effective selective breeding of red sea bream to purge the causative gene of the undesirable phenotype and improve seed production of red sea bream as well as provide basic information of the mechanisms of thyroid hormones and its related diseases in fish and humans.
Similar content being viewed by others


Introduction
With the worldwide expansion of the aquaculture industry (FAO, 2019), fish are increasingly selectively bred for rapid growth and disease resistance, creating more robust strains to expand production (Houston et al., 2020; Teletchea and Fontaine, 2014). With few exceptions, fish farming has a shorter history than livestock and crops, some of which have been bred over thousands of years. Currently, 30 fish species have been subjected to selective breeding programs following a path similar to that in terrestrial livestock (Teletchea and Fontaine, 2014). Rapidly growing disease-resistant strains have been developed for aquaculture (Gjedrem, 2015; Lhorente et al., 2019). Domestication involves selective breeding of successive generations using a limited number of broodstock, consequently reducing the effective population size and accelerating inbreeding (Frantz et al., 2020; Groeneveld et al., 2010; Larson and Burger, 2013).
The red sea bream Pagrus major, as its common name suggests, is bright red. Because red is a symbol of happiness in Japan, this fish is traditionally consumed at auspicious events such as birth and wedding celebrations, making body color an important factor in its economic value (Foscarini, 1988). Red sea bream farming is now an important sector of the Japanese fishery industry, where it has been selectively bred for fast growth (Murata et al., 1996; Sawayama and Takagi, 2015) and disease resistance (Sawayama et al., 2019a, 2019b) over the past half-century (Sawayama and Takagi, 2016). Approximately 50 million P. major seedlings are artificially produced annually for rearing on farms in Japan (Makino, 2017). The entire lifecycle of farmed red sea bream has been artificially controlled for over ten generations, dramatically decreasing its effective population size compared to that of the wild stock (Sawayama and Takagi, 2016). Inbreeding is accelerating in cultured populations and may give rise to negative effects, including genetic anomalies such as bone malformation and atypical/incomplete body color (Sawayama et al., 2014; Sawayama and Takagi, 2011; Sawayama and Takagi, 2012). Identifying genes inducing such abnormalities is necessary to improve production and maintain animal welfare.
We previously reported a novel body color abnormality in artificially produced juvenile red sea bream (Sawayama and Takagi, 2011). Blood vessels, vertebrae, and bones of the abnormal individuals are clearly visible, so we designated it the transparent (TP) phenotype. The TP phenotype is the result of delay in chromatophore development compared with that in fish exhibiting a normal body color (NC) phenotype (Sawayama and Takagi, 2011). The TP phenotype gradually acquires some body color after 40 dph, but remains pale and readily distinguishable from the NC phenotype. It is known that chromatophore development related to body color expression is controlled by thyroid hormones (Hirata et al., 1989; Trijuno et al., 2002), and a thyroid hormone surge occurs between 30 and 40 dph at the larva-to-juvenile transition in red sea bream (Kimura et al., 1992). Hence, the red sea bream TP phenotype is suggested to be a thyroid hormone-related disorder. Using amplified fragment-length polymorphism analysis, we recently identified a novel single-nucleotide polymorphism (SNP) significantly associated with the TP phenotype. The genotype patterns of the TP-specific SNPs suggest that the phenotype follows simple Mendelian inheritance (Sawayama et al., 2018). However, observations of the frequency of the TP phenotype at 40 dph are significantly lower than would be expected (Sawayama and Takagi, 2011), suggesting high mortality at early stages. This, along with the undesirable body color of survivors, could imply loss of ~25% of offspring from a pair exhibiting the mutant allele. This is a significant cost to commercial red sea bream seed production that could be ameliorated through selective breeding to eliminate the deleterious gene.
Our objective was to identify the candidate gene responsible for the TP phenotype using family-based linkage mapping. Candidate genes were further genotyped in several farmed populations. Observations of physiology and morphology, a rescue experiment, and CRISPR–Cas9-mediated loss-of-function analysis were conducted to support evidence of the TP phenotype candidate gene.
Materials and methods
Family construction for linkage mapping
One red sea bream male exhibiting the TP-specific SNP was obtained from a seed production company (Marua Suisan Co., Ltd., Ehime, Japan) and used to fertilize naturally spawned eggs from 20 females (G0 population) (Sawayama et al., 2019a). After two years of culture, twenty G1 specimens were genotyped as TP-360, and nine females and a single male were confirmed heterozygous for the TP-specific allele.
A pair with the TP-specific SNP was selected from the G1 population, and F1 was produced by artificial fertilization. The offspring were reared in a 500-l tank following standard hatchery practice. At 33 dph, the phenotypes were visually distinguishable. All specimens were euthanized by overdose of 2-phenoxyethanol and stored in 99.9% ethanol until DNA extraction. The number of samples and phenotype frequency in the experimental family are shown in Table S1.
Genotyping for linkage mapping
Genomic DNA from NC (n = 96) and TP phenotypes (n = 96) was extracted using the QuickGene DNA tissue kit (KURABO, Osaka, Japan). Double-digest RAD-seq (ddRAD-seq) was conducted based on Peterson et al., (2012). Briefly, genomic DNA was digested by MseI and EcoRI restriction enzymes. Adapters were ligated to the digested DNA, and size-selected (500 ± 50 bp) using LabChipXT (PerkinElmer, Billerica, MA, USA). The DNA fragments were amplified by PCR for 12 cycles. After quantification and qualification, the library was sequenced with single-end 76 bp using NextSeq 500 (Illumina, California, USA).
The Illumina raw reads were demultiplexed to each specimen based on 8 bp of dual-index sequences. Quality filtering was conducted using FASTX Toolkit v. 0.0.14 with the function fastq_quality_trimmer (Phred score < 20, length < 30 bp). The trimmed sequences were mapped to red sea bream reference genome (GCA_002897255.1) using Bowtie2 v. 2.2.6. (Langmead et al., 2019), and a BAM file was generated using SAMtools v. 1.3 (Li, 2011). Single-nucleotide polymorphisms were extracted using SAMtools, and the data format was converted to VCF using BCFtools. Quality filtering was conducted using VCFtools (Danecek et al., 2011) set to minimum quality 10, minimum depth 10.
We used 71 microsatellite markers to compare the linkage groups constructed by Sawayama et al. (2017). All procedures for microsatellite analysis followed Sawayama et al. (2017).
Linkage mapping and candidate gene mining
A linkage map was constructed using JoinMap v. 3 (Kyazma, Wageningen, Netherlands) using 88 progeny (44 NC and 44 TP) and the parent fish. Genotype data were treated as outbreeder full-sib family (CP) for linkage map construction. We included SNPs genotyped in over 90% of specimens and omitted those distorted from the Mendelian ratio (P < 0.05), and high-quality SNPs were used for linkage map construction. Sex-specific linkage maps were constructed and integrated into a consensus map. A marker with a logarithm of odds threshold ≥ 5.0 was assigned to the linkage group. Graphic map files were generated using MapChart v. 2.2 (Voorrips, 2002). Genome length was estimated in the consensus maps according to the method of Fishman et al. (2001).
Genotype data were subsequently converted to F2 format for further linkage analysis. Genome-wide linkage analysis was performed using J/qtl software (Smith et al., 2009), the graphic interface of R/qtl (Broman et al., 2003), by implementing the binary model to test 10,000 permutations of the genotype vector generated for each parent using standard interval mapping (expectation-maximization likelihood method). Single quantitative trait loci were scanned by the scanone function of the R/qtl package across all linkage groups.
All specimens (96 NC and 96 TP) were used for fine mapping of the specific LG for which significant linkage was observed. No filtering by Mendelian ratio was conducted because the SNPs significantly linked to the TP phenotype were distorted from the Mendelian ratio in the TP group.
The scaffold containing the SNPs perfectly linked to the TP phenotype was searched in GenBank using BLAST to identify possible candidate genes.
Identifying nucleotide mutations in candidate genes
Forty-eight 20 dph larvae produced by the seed production company (Marua Suisan Co., Ltd.) in 2016 were randomly collected for screening SNPs in the candidate genes. The head was removed with scissors and stored at −80 °C with RNAlater (Thermo Fisher Scientific, Waltham, MA, USA) for later analysis. The remainder of the body was stored in 100% ethanol and subjected to TP-360 genotyping (Sawayama et al., 2018) to establish phenotype. Juveniles inheriting A/A and A/G genotypes were classified as NC and those with the G/G genotype as TP. Two individuals of the A/A genotype and two of G/G genotype were randomly selected and used for further experimentation.
Coding regions of the candidate gene were predicted from the red sea bream scaffold (GCA_002897255.1) by Splign (Kapustin et al., 2008) using medaka cDNA sequences, and primer pairs were designed to amplify mRNA of the candidate gene using Primer3 (Rozen and Skaletsky, 2000). Total RNA was extracted using ISOGEN with the Spin Column kit (NipponGene, Tokyo, Japan) from the head of 20 dph larvae selected as described. Complementary DNA was synthesized using PrimeScript II 1st strand cDNA synthesis kit (Takara Bio, Shiga, Japan) according to the manufacturer’s instructions. The PCR was conducted with 1 µl of cDNA solution, 0.3 µM primer, 0.4 µM dNTPs, and 0.2 units of KOD Fx polymerase (Toyobo, Osaka, Japan) with 2× buffer at a total volume of 10 µl. The PCR reactions consisted of an initial denaturing step for 2 min at 94 °C followed by 30 cycles of 98 °C for 10 s, 60 °C for 30 s, and 68 °C for 30 s. The PCR products were cloned into pGEM T-easy vector (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Cycle sequencing was performed using a BigDye Terminator v. 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). All samples were sequenced on an ABI PRISM 3130xl genetic analyzer (Applied Biosystems). Obtained nucleotide sequences were aligned with MEGA 6 software (Tamura et al., 2013) using the ClustalW algorithm. Nucleotide sequences were converted to amino acid residues using MEGA 6, and SNPs causing amino acid changes were identified. Because we did not clone full-length cDNA of target genes, the location of the SNP was shown as the position of scaffold. Identified SNPs were evaluated for further analysis.
Association analysis
To clarify the association of the mutations of possible candidate genes with phenotype, we obtained NC and TP phenotype seedlings generated in 2012 and 2015 from four commercial broodstock populations (Sawayama et al., 2018). Fish of NC (n = 171) and TP (n = 165) phenotype from the four production lots were used for genotype candidate loci and association analysis. All DNA samples were stored at −20 °C until genetic analysis. PCR and direct sequencing were conducted as described. Association between phenotype and genotype was evaluated using Haploview (Barrett et al., 2005). Linkage disequilibrium among SNPs was calculated and illustrated schematically using Haploview.
Protein structure analysis of the candidate gene
The transmembrane structure of candidate genes plays a critical role in protein function, hence, we evaluated whether transmembrane structure might be affected by TP phenotype–specific mutations. Transmembrane structures of candidates were analyzed using SOSUI (Hirokawa et al., 1998) and TMHMM (Krogh et al., 2001) with default parameter settings.
Characterization of the TP phenotype
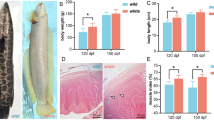
Erythrocytes of NC and TP phenotypes were compared at 40, 45, 50, 60, 70, 80, and 90 dph. The tail was cut from the body under anesthesia by 0.05% 2-phenoxyethanol, and a drop of blood was obtained (n = 3/phenotype each sampling day). The blood was smeared, air-dried, fixed in 99% methanol, and stained with May–Grünwald/Giemsa, and one-hundred erythrocytes per slide examined by light microscopy (Nikon, Tokyo, Japan) at 1000×. Length, width, and area of erythrocytes and their nuclei were measured using NIS-Element software (Nikon). Erythrocyte area (AE), nucleus area (AN), nucleus area in erythrocyte (NAE, %), ratio of long/short diameter of erythrocyte (E_L/S), and ratio of long/short diameter of nucleus of erythrocyte (N_L/S) were recorded.
Thyroxine (T4) was measured in NC and TP phenotypes at 35, 40, 45, 50, 60, 70, 80, and 90 dph. Whole body was homogenized in cold ethanol (99.9%) and the supernatant was isolated after centrifugation at 1600 × g at 4 °C. The supernatant was dried using a centrifugal evaporator at 37 °C, and 1× PBS (pH 8.6) with a 10% volume of 99% ethanol was added and vortexed. The extracts were stored at −40 °C until further analysis. Total T4 was measured with T4 ELISA kit (Immunospec Corporation, California, USA) using microplate reader Infinite F200 PRO (Tecan, Zürich, Switzerland).
The thyroid gland was examined in 50 dph NC and TP fish (n = 3/phenotype) when metamorphosis in the NC phenotype was complete. The head was fixed with Bouin’s solution and dehydrated using ethanol and xylene. The dehydrated tissue was embedded in paraffin and cut longitudinally into 5 µm sections. Hematoxylin & eosin staining was conducted according to standard methods, and the sections were examined under a microscope (BX51, Olympus, Japan).
Phenotype rescue
Transparent phenotype fish were collected at 35 dph from a commercial production lot and reared in 10-l plastic tanks in 2016. The numbers of chromatophores increased with fish growth in the TP phenotype (Sawayama and Takagi, 2011), suggesting an effect of endogenous T4. Therefore, to determine whether the transparent phenotype was rescued by exogenous thyroid hormone, TP fish were treated with thiourea to inhibit endogenous T4 synthesis. Treatment was administered according to Hirata et al. (1989) and Trijuno et al. (2002). After five days of acclimation, the following agents were added to the water to create three experimental groups: 30 mg/l thiourea (TU) (Nacalai Tesque Co., Ltd., Kyoto, Japan) dissolved directly in seawater, 0.1 mg/l L-thyroxine sodium salt (T4; Nacalai Tesque Co., Ltd.) dissolved in 99.5% ethanol, and 10 µl/l of 99.5% ethanol as control. Ten TP fish were introduced to each experiment tank. Half the tank water was replaced each day, keeping the chemical concentrations constant. The experiment was terminated after five days, and five surviving fish from each experimental group were collected and photographed under a stereomicroscope (SZ61, Olympus, Japan), and a morphological examination conducted. Normal-color fish at 40 and 45 dph (n = 10 for each dph) were also collected from the same commercial production lot, and examined as for the TP fish. The a* value of LAB color space, which indicates redness, was measured using NIS-Element software (Nikon) for ~6 mm2 of body surface and normalized over the area (a* value/mm2). The normalized a* value of each group was compared with one-way ANOVA followed by Tukey’s test using the statistics package Past 4 (Hammer et al., 2001).
Loss of function of the candidate gene
To evaluate whether the mutation found in the red sea bream TP phenotype is the source of the transparency, CRISPR/Cas9 genome-editing of duoxa (XM_002666907.5) was conducted using zebrafish Danio rerio. Red sea bream requires a long period for maturation (usually three years in female) and large facilities for maintenance. Therefore, we used zebrafish, which is small with a shorter life cycle. The target sequence of guide RNA (gRNA) was designed on exon 3 of the gene using CRISPRdirect (Naito et al., 2015), and off-target sites were searched for mismatch 4, DNA bulge 0, and RNA bulge 0 using Cas-OFFinder (Bae et al., 2014). Finally, twenty nucleotide sequences, CAAATCCTTCAGCACGGTGG, were selected for gRNA with the protospacer-adjacent motif (TGG) at the 3´ end. A combination of 50 ng/µL of crRNA (Integrated DNA Technologies, Coralville, IA, USA) and 50 ng/µL of tracrRNA was microinjected into 1- and 2-cell stages of wild-type zebrafish eggs with Cas9 nuclease (Alt-R® S.p. Cas9 nuclease 3NLS; Integrated DNA Technologies). Injected eggs were incubated at 28.5 °C until hatching. CRISPR/Cas9-mediated genome editing might induce homozygous mutation in duoxa, leading to dramatically reduced survival in F0 larvae. To increase survival, F0 larvae were reared in water containing 0.1 mg/l L-thyroxine sodium salt until metamorphosis was complete. The phenotype of F0 individuals was determined visually, and transparent-like individuals were selected and used for F1 production. The F1 individuals were reared to ~2 cm total length, and the phenotype was determined. The exon 3 of duoxa in crispants was cloned and sequenced, and nucleotide and amino acid sequences of duoxa in crispants were identified. Wild-type zebrafish were used for comparisons of phenotypes and nucleotide sequences.
Results
Linkage map construction
A total of 194 ddRAD-seq libraries from parents and 192 offspring (NC = 96 and TP = 96) were constructed and sequenced on Illumina NextSeq 500 platform to generate 287.1 million raw reads (Table S2). After trimming and filtration, the reads were mapped to the reference genome of red sea bream (GCA_002897255.1). Sequencing data of eight NC specimens were omitted from further analysis because of an insufficient number of reads.
For linkage map construction, we selected 88 high-quality–genotyped individuals (NC = 44 and TP = 44) along with the parent fish, and 5365 loci were obtained. The loci were filtered for genotype rate and distortion from the Mendelian ratio, and 2570 loci were selected as high-quality SNPs (Table S3). The selected loci and microsatellites were used for linkage map construction, and 1183 SNPs and 37 microsatellites were mapped to 25 and 26 linkage groups in female and male, respectively (Fig. S1). A summary of marker segregation patterns and linkage maps is presented in Tables S4 and S5, respectively. Maternal maps contained 993 loci with a total genetic distance of 1056.6 cM. Paternal maps contained 853 loci with a total genetic distance of 804.5 cM. The length of the maternal map was 1.49 times that of the paternal map. Consensus maps contained 1220 loci with a total genetic distance of 1201.2 cM. The total bps of mapped contigs/scaffolds was approximately 15.1 Mbp (1.7% of assembled sequences). The genome coverage of the consensus maps was estimated at 95.7%.
Screening for candidate LG and candidate genes
All markers located on LG23 were significantly linked to the TP phenotype (Fig. 1A). Therefore, the candidate gene(s) was suggested to be located on this LG, especially near the markers with the highest LOD score. LG23 was also identified as the location of the TP-specific SNP in Sawayama et al., (2018).

A Genome-wide linkage analysis across 24 female-male consensuses linkage groups with one female- and two male-specific linkage groups. Each linkage group is separated by a vertical dotted line with a number on the x-axis. Log-of-odds scores are plotted on the y-axis. B Chromosome-wide analysis of the TP phenotype after fine-mapping LG23. Marker location (cM) and log of odds scores are shown on x- and y-axes, respectively. The horizontal dotted lines indicate genome-wide significance cutoffs of log-of-odds scores (P < 0.05).
Filtered SNPs diverging from the Mendelian ratio were remapped to LG23, and 146 markers were mapped onto the LG (Fig. 1B and Table S6). Length of the remapped LG23 was 95.8 cM. The SNPs located on two LG23-composing scaffolds, scaffold219537.6 (BDUH01021953.1: 37,010 bp) and scaffold640417.9 (BDUH01064041.1: 235,943 bp), were perfectly linked to the TP phenotype (n = 184). Nucleotide sequences of the two scaffolds were searched by BLASTn. No gene was found in scaffold 640417.9, but two genes, dual oxidase (duox) and dual oxidase maturation factor (duoxa) were found in scaffold 219537.6. These genes are well-known to be involved in thyroid hormone synthesis (Grasberger, 2010). No other genes were found in scaffold219537.6.
Screening of nucleotide mutations in candidate genes
Thirty exons were predicted from red sea bream duox based on medaka genome data. Eight primer pairs were designed for screening nucleotide mutations on exons of red sea bream duox (Table S7). Twenty-three SNPs and one deletion were found in duox mRNA (LC627593) with four identified as missense mutations (nucleotide positions 1193, 1194, 1710, and 2505 in LC627593). These SNPs were designated duox_SNP1–SNP4. The SNPs duox_SNP1 (g.36377G > A) and duox_SNP2 (g.36376C > G) were adjacently located in exon 8. The combination of the two SNPs mutated Ala to Gly, Ser, or Thr. In exon 12 of duox, duox_SNP3 (g.35435T > C) mutated Ala to Val. duox_SNP4 (g.31956G > A) in exon 16 of duox mutated Lys to Arg. These SNPs were used for further association analysis.
Six exons were predicted in red sea bream duoxa based on medaka genome data, and the length of the coding region was estimated as 870 bp (Fig. 2A). We designed three primer pairs to amplify six exons from genomic DNA of red sea bream for SNP screening (Table S7). Two SNPs were found on exon 3 of the gene, and one SNP (duoxa_SNP, g.44310G > A) was nonsynonymous and mutated Gly to Arg (Fig. 2B–D). This mutation was confirmed using cDNA obtained from larvae inheriting TP and NC genotypes of TP-360. The partial duoxa mRNA sequences (917 bp) were deposited in GenBank (LC627592). The SNPs were located at 267 bp and 268 bp. The position of missense mutation was at 51 amino acid residue (p.Gly51Arg). We designed a new primer pair to amplify and genotype the missense mutation (Table S7) for further association analysis.

A Position of SNPs in the gene. Box and line represent exon and intron, respectively. Arrow on exon 3 indicates SNPs observed in TP phenotype. B–D SNPs and allelic patterns obtained by direct sequencing. Two SNPs are shown in the red boxes, distributed over two codons. One SNP (T or A), the 3´ end of Val, does not change the amino acid. G or T SNP, the 5´ end of Gly, changes the amino acid Gly to Arg. Electropherogram showing heterogeneous alleles is in C.
Association analysis
A total of 161 NC and 165 TP fish from different production lots were genotyped by the obtained five nonsynonymous SNPs, and data were used for association analysis. All SNPs showed significant association with the TP phenotype (P < 0.05), with the most significant P-value (97.2 as −log10-transformed) observed in duoxa_SNP (Fig. 3A). A seven kb haplotype block was estimated between duox_SNP2 and duoxa_SNP (Fig. 3B). Genotype frequencies of the five nonsynonymous SNPs are shown in Table 1. All loci within duox were significantly associated with the TP phenotype, and the duoxa_SNP showed 100% association with TP. All NC individuals inherited GG (62.7%) or AG (37.3%), while all TP individuals inherited AA.

A Plot of -log10 (P-value) of four SNPs of duox and one SNP of duoxa. The position of each SNP on scaffold219537.6 (235,943 bp) is shown on x-axis. Each position is connected to the schematic diagram of Fig. 3B. B Linkage disequilibrium among two genes. The square of the correlation coefficient (r2) is estimated for each pairwise comparison of SNPs, and r2 value of each pairwise comparison is shown in each box. The estimated haplotype block, the 7 kb region, including duox_SNP2, duox_SNP1, and duoxa_SNP, is shown by the inverted triangle.
Transmembrane structure of DUOXA
Since structure affects protein function, we evaluated the mutation’s impact on the transmembrane structure of DUOXA (Grasberger, 2010). Four primary (amino acid residues 19–41, 52–74, 180–202, and 249–271) and two secondary (amino acid residues 92–113 and 213–235) transmembrane structures were predicted in wild-type DUOXA, but the secondary structure observed in amino acid residues 92–113 was not predicted in mutant DUOXA by the SOSUI program. The TMHMM method predicted transmembrane structures similar to SOSUI (Fig. 4). Five transmembrane structures were predicted in both wild (Fig. 4A) and mutant DUOXA (Fig. 4B). The amino acid residues from 90–112 showed moderate probabilities of the transmembrane helix (0.149 to 0.358) in wild type (Fig. 4A). The probability of the outside loop was high (> 0.9) before the amino acid residue 90, but this value was then decreased until 0.6 by the next transmembrane helix in wild type. The probability of the inside loop was low at the amino acid residues from 90 to 112 but then increased to around 0.4 by the next transmembrane helix in wild type. No such structure was found in that region in the mutant DUOXA (Fig. 4B). This region was also suggested as a secondary transmembrane structure in SOSUI. No structural change resulting from the four missense mutations in DUOX (data not shown) was identified using TMHMM and SOSUI.

A Wild type, and B mutant of DUOXA. Red bars indicate the probability of a transmembrane helix, and blue and magenta lines indicate the probability of intracellular and extracellular loops.
Phenotypic evaluation of TP individuals
To corroborate candidate genes identified by genetic analysis, several phenotypic traits were assessed. Nuclei of erythrocytes from the TP phenotype appeared more circular than those in the NC phenotype (Fig. S2). Morphometric analysis of erythrocytes confirmed the results of visual examination, revealing significant differences in AN, NAE, and N_L/S at 40 dph (P = 0.006, 0.003, and 0.002, respectively) of TP and NC. The N_LS difference remained significant at 45 dph (P = 0.006) (Fig. S3).
Histological analysis revealed few thyroid follicles in the NC phenotype (Fig. S4A, B), while a large number of thyroid follicles were observed in the area of the esophagus and lower jawbones in TP fish (Fig. S4C, D). The mean (SD) number of thyroid follicles in three NC and TP specimens was 17.0 (0.8) and 83.3 (15.2), respectively, significantly differing (P < 0.05) by the Mann–Whitney U test.
Total T4 concentration differed significantly in NC and TP fish at 35 dph (P < 0.001), but no significant difference was observed on other sampling days (Fig. S5).
Phenotype rescue
The survival rate of fish in the five-day rescue experiment was 90% in control, 70% in the T4 group, and 60% in the TU group. The main cause of mortality in T4 was cannibalism but, in TU, no clear cause was identified. The NC and TP phenotypes at the beginning of the experiment (40 dph) are shown in Fig. 5A–D. Stripes were visible in the NC phenotype (Fig. 5A), and many small melanophores were observed on the body surface (Fig. 5B). No stripes were present in the TP phenotype (Fig. 5C), and a small number of large melanophores were observed (Fig. 5D) as previously reported (Sawayama and Takagi, 2011).

A Initial body color of NC and B its magnified body surface at 40 dph.C Initial body color of TP and D its magnified body surface at 40 dph. E NC phenotype and F its magnified body surface at 45 dph. G–L Typical TP individuals after 5 days of rescue experiment (45 dph). G TP phenotype treated with ethanol and H its magnified body surface, I TP phenotype treated by 0.1 mg/l of thyroxin (T4) and J its magnified body surface, K TP phenotype treated by 30 mg/l of thiourea (TU) and L its magnified body surface. Scale bars for whole body and magnified body surface represent 1 mm and 10 μm, respectively.
Fish after five days of treatment are shown in Fig. 5E–L. The red color of the NC phenotype appeared more intense than in the previous five-day period (Fig. 5E, F). The number of melanophores at 45 dph in the TP control group was higher than observed in 40 dph TP fish. Stripes were similar to those of NC (Fig. 5G), but melanophores were larger (Fig. 5H). The number of melanophores was higher in T4-treated TP fish (Fig. 5I) than in the TP control group. Melanophores were smaller than in TP controls and similar to those of normal fish (Fig. 5J). The TU-treated TP fish showed a phenotype similar to the TP control group (Fig. 5K, L).
The NC fish appeared reddish, while TP fish were transparent at 40 dph. This was confirmed by the a* value at 40 dph (Fig. S6A). The T4-treated TP fish showed less intense red color than the NC phenotype of 45 dph, but more than TU-treated and TP control fish (Fig. S6B).
CRISPR–Cas9-mediated loss-of-function
The duoxa crispants (−/−) were obtained by breeding zebrafish of the F0 generation heterozygous for CRISPR–Cas9-edited duoxa (+/−). The wild type fish showed clear vertical lines on the body surface as well as many melanophores (Fig. 6B). Internal structures were not visible (Fig. 6A). The F1 crispants showed a transparent-like phenotype, the vertical lines on the body surface were indistinct, and internal organs were visible (crispant #1, Fig. 6C; crispants #2–5, Fig. S7A–D). Crispants exhibited fewer and smaller melanophores than those in the wild type (crispant #1, Fig. 6D). The phenotypic features of the zebrafish F1 crispants were similar to the TP red sea bream phenotype. The thyroid gland of the crispants was examined, but no notable differences were observed between crispants and wild type (data not shown).

A Whole body of wild type zebrafish and (B) its body surface. C Whole body of the F1 crispant and (D) its body surface. Scale bars in (A, C) represent 10 mm. Scale bars in (B, D) represent 1 mm.
All F1 crispants exhibited truncated and/or inserted nucleotides of exon 3 (Fig. S8A), and some induced nonsense mutations (Fig. S8B alleles −17 and +15−3).
Discussion
A transparent phenotype of juvenile red sea bream is often observed in seed production, and previous research has suggested simple Mendelian inheritance (Sawayama and Takagi, 2011; Sawayama et al., 2018). Theoretically, 25% of offspring from a broodstock pair inheriting a mutant allele will exhibit the TP phenotype, and this directly induces a production loss. Hence, there is a significant demand to identify the causative gene of the TP phenotype in red sea bream seed production.
The presented linkage map contains 1220 loci (1183 SNPs and 37 microsatellites) and covers 95.7% of the red sea bream genome, expanding the number of mapped loci from 192 to 1220 and covering ~1.5 times the genome of the previous map (Sawayama et al., 2017). However, only 1.7% of the assembled sequences were mapped, and some LGs still showed a small number of mapped loci, including LG21 (n = 4) to LG24 (n = 14), with low genetic length. In addition, the number of linkage groups (n = 26) exceeded the number of chromosomes in the red sea bream (n = 24). Domestic red sea bream used in this study was the product of more than 10 generations, and the mapping family was highly inbred. Therefore, the power to detect recombination events was lower. Generating a test cross using individuals from different breeding lines or wild individuals will increase loci-mapped and linkage-grouping accuracy. In addition, the previously assembled genome of red sea bream contained a large number of scaffolds (n = 886,260). Recently, a red sea bream reference genome information generated from a long-read sequencer was provided (Shin et al., 2018) that will be useful to expand linkage groups to the pseudo-chromosome level.
Loci significantly linked to the TP phenotype were mapped on LG23, where the TP-specific SNP was mapped in Sawayama et al. (2018). Two genes, duox and duoxa, related to thyroid hormone synthesis, were identified as candidate genes by the genome-wide and chromosome-wide linkage analyses. They are homologs of duox2 and duoxa2 in mammals, birds, and amphibians (Grasberger and Refetoff, 2006) that generate H2O2 at the apical membrane of thyroid follicular cells and are essential for iodination in thyroid hormone synthesis (Grasberger and Refetoff, 2006). Heterodimeric complexes of DUOX and DUOXA generate H2O2, and thyroid peroxidase utilizes H2O2 as the essential substrate for iodide oxidation and iodination of thyroglobulin (Grasberger, 2010; Weber et al., 2013). Mutations of both genes cause congenital hypothyroidism in humans (Hulur et al., 2011; Liu et al., 2015; Yi et al., 2013; Zamproni et al., 2008; Zheng et al., 2016).
We found only one missense mutation in duoxa (Fig. 2), which was perfectly associated with the TP phenotype (Table 1). In addition, in silico analysis of DUOXA transmembrane structure suggests that the mutation affects the secondary structure formation of transmembrane (Fig. 4). This secondary structure is observed in DUOXA of normal red sea bream and zebrafish, and its absence may influence the gene function. No mutations in the same region of duoxa have been reported in humans (Hulur et al., 2011; Yi et al., 2013; Zamproni et al., 2008; Zheng et al., 2016), and it is not clear how this mutation affects H2O2 generation in the thyroid gland.
Similar to the duoxa2 mutation, mutation of duox2 induces congenital hypothyroidism in mammals, including humans (Cao et al., 2019; Kizys et al., 2017). In addition, crispants of duox have shown pigmentation defects and thyroid hyperplasia/external goiter in zebrafish (Chopra et al., 2019), and hyperplastic goiter was more frequently observed in duox crispants than in duoxa crispants in zebrafish (Trubiroha et al., 2018). Similar to zebrafish duox crispants (Chopra et al., 2019), an enlarged thyroid gland was found in the red sea bream TP phenotype (Fig. 5C, D). In addition, significant association with the TP phenotype was observed in duox_SNP1, which is located in the same haplotype block as duoxa_SNP (Fig. 3B). This suggests mutations in both duox and duoxa as possible sources of the TP phenotype, and missense mutations of duox observed in this research may be related to the TP phenotype of red sea bream. However, duox_SNP1 was not perfectly associated with the TP phenotype in fish from different production lots (Table 1). This implies that missense mutations in duox are not the primary source of the TP phenotype in red sea bream, but that mutation of duox is possibly a factor in goiter formation in the TP phenotype of red sea bream. Further genome editing of duox will be required to confirm goiter formation in red sea bream.
In addition to atypical body color and thyroid gland morphology, thyroid disfunction was also suggested based on several phenotypic features of TP fish. The shape of red blood cell nuclei changed between 40 and 45 dph (Fig. S4). Erythrocytes of 40 dph TP fish showed large circular nuclei, similar to immature erythrocytes in fish (Miwa and Inui 1991; Inui et al., 1995). The TP fish exhibited mature erythrocytes at ~50 dph. The TP has also been reported to gradually assume more normal body color after 40 dph (Sawayama and Takagi, 2011). These data suggested that T4 sufficient for normal development is produced by some detour mechanism after 40 dph. The T4 concentration of the TP phenotype also supports existence of detour mechanisms. The T4 concentration of 35 dph TP fish was significantly lower than seen in the NC phenotype, but concentration gradually increased and showed a pattern similar to the NC phenotype at 40 dph.
As previously stated, the DUOX/DUOXA heterodimer generates H2O2 for thyroid hormone synthesis (Grasberger, 2010; Weber et al., 2013), but DUOX can generate some H2O2 without DUOXA (Grasberger and Refetoff, 2006). The T4 concentration in the TP red sea bream phenotype increased after 40 dph (Fig. S5); therefore, assimilation of endogenous T4 may be recovered by DUOX or by an alternative pathway at ~40 dph in TP phenotype. Transient congenital hypothyroidism caused by duox2 is observed in humans (Maruo et al., 2008), and further research will be required to identify the mechanisms of T4 recovery associated with the duoxa mutation in the TP phenotype of red sea bream.
The mutation of the duoxa gene found in this study will provide a useful tool for selecting red sea bream broodstock without the deleterious mutation. The TP phenotype follows simple Mendelian inheritance; thus 25% of offspring of a pair exhibiting the mutant allele will hypothetically express the TP phenotype. Marker-based selection is able to purge the causative gene of the TP phenotype and improve seed production of red sea bream.
Over decades, a large number of farmed red sea bream have escaped in Japan, and crossing of farmed with wild fish has occurred (Sawayama et al., 2019b). The deleterious mutation in farmed fish has been transmitted to wild populations, possibly influencing their development and survival. The identified mutation of duoxa will aid in the direct evaluation of the impact of domestic fish escapes on natural populations.
Some genetic deformities in farmed fish seem related to larva-to-juvenile transition (Sawayama et al., 2014), and congenital hypothyroidism may be one of the major genetic deformities in cultured fish populations. The present research provides evidence of the usefulness of genome-wide scale linkage analysis. Developing and improving reference genome information of farmed fish species will benefit from identifying useful DNA markers and causative genes of genetic disorders. Understanding genetic disorders of farmed fish species will improve production and animal welfare, and this study also provides basic information of the mechanisms of thyroid hormone and its related disease in vertebrates, including fish and humans.
References
Bae S, Park J, Kim J-S (2014) Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30:1473–1475
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Cao C, Zhang Y, Wang X, Zheng Q, Zhang H, Song R et al. (2019) An exonic splicing enhancer mutation in DUOX2 causes aberrant alternative splicing and server congenital hypothyroidism in Bama pigs. Dis Model Mech 12:dmm036616
Chopra K, Ishibashi S, Amaya E (2019). Zebrafish duox mutations provide a model for human congenital hypothyroidism. Biol Open 8:bio037655
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
FAO (2019). FAO yearbook. Fishery and aquaculture statistics 2017. http://www.fao.org/fishery/statistics/yearbook/en
Fishman L, Kelly AJ, Morgan E, Willis JH (2001) A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics 159:1701–1716
Foscarini R (1988) A review: Intensive farming procedure for red sea bream (Pagrus major) in Japan. Aquaculture 72:191–246
Frantz LAF, Bradley DG, Larson G, Orlando L (2020) Animal domestication in the era of ancient genomics. Nat Rev Genet 21:449–460
Gjedrem T (2015) Disease resistant fish and shellfish are within reach: a review. J Mar Sci Eng 3:146–153
Grasberger H (2010) Defects of thyroidal hydrogen peroxide generation in congenital hypothyroidism. Mol Cell Endocrinol 322:99–106
Grasberger H, Refetoff S (2006) Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem 281:18269–18272
Groeneveld LF, Lenstra JA, Eding H, Toro MA, Scherf B, Pilling D et al. (2010) Genetic diversity in farm animals–a review. Anim Genet 41:6–31
Hammer Ø, Harper DA, Ryan PD (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron 4:9
Hirata Y, Kurokura H, Kasahara S (1989) Effects of thyroxine and thiourea on the development of larval red sea bream Pagrus major. Nippon Suisan Gakkaishi 55:1189–1195
Hirokawa T, Boon-Chieng S, Mitaku S (1998) SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379
Houston RD, Bean TP, Macqueen DJ, Gundappa MK, Jin YH, Jenkins TL et al. (2020) Harnessing genomics to fast-track genetic improvement in aquaculture. Nat Rev Genet 21:389–409
Hulur I, Hermanns P, Nestoris C, Heger S, Refetoff S, Pohlenz J et al. (2011) A single copy of the recently identified dual oxidase maturation factor (DUOXA) 1 gene produces only mild transient hypothyroidism in a patient with a novel biallelic DUOXA2 mutation and monoallelic DUOXA1 deletion. J Clin Endocrinol Metab 96:E841–E845
Inui Y, Yamano K, Miwa S (1995) The role of thyroid hormone in tissue development in metamorphosing flounder. Aquaculture 135:87–98
Kapustin Y, Souvorov A, Tatusova T, Lipman D (2008) Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct 3:20
Kimura R, Tagawa M, Tanaka M, Hirano T (1992) Developmental changes in tissue thyroid hormone levels of red sea bream Pagrus major. Nippon Suisan Gakkaishi 58:975
Kizys MML, Louzada RA, Mitne-Neto M, Jara JR, Furuzawa GK, de Carvalho DP et al. (2017) DUOX2 mutations are associated with congenital hypothyroidism with ectopic thyroid gland. J Clin Endocrinol Metab 102:4060–4071
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Langmead B, Wilks C, Antonescu V, Charles R (2019) Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35:421–432
Larson G, Burger J (2013) A population genetics view of animal domestication. Trends Genet 29:197–205
Lhorente JP, Araneda M, Neira R, Yáñez JM (2019) Advances in genetic improvement for salmon and trout aquaculture: the Chilean situation and prospects. Rev Aquac 11:340–353
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993
Liu S, Liu L, Niu X, Lu D, Xia H, Yan S (2015) A novel missense mutation (I26M) in DUOXA2 causing congenital goiter hypothyroidism impairs NADPH oxidase activity but not protein expression. J Clin Endocrinol Metab 100:1225–1229
Makino M (2017). National Aquaculture Sector Overview. Japan. National Aquaculture Sector Overview Fact Sheets. http://www.fao.org/fishery/countrysector/naso_japan/en
Maruo Y, Takahashi H, Soeda I, Nishikura N, Matsui K, Ota Y et al. (2008) Transient congenital hypothyroidism caused by biallelic mutations of the dual oxidase 2 gene in Japanese patients detected by a neonatal screening program. J Clin Endocrinol Metab 93:4261–4267
Miwa S, Inui Y (1991) Thyroid hormone stimulates the shift of erythrocyte populations during metamorphosis of the flounder. J Exp Zool 259:222–228
Murata O, Harada T, Miyashita S, Izumi K-I, Maeda S, Kato K, Kumai H (1996) Selective breeding for growth in red sea bream. Fish Sci 62:845–849
Naito Y, Hino K, Bono H, Ui-Tei K (2015) CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31:1120–1123
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7:e37135
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Sawayama E, Asahina K, Takagi M (2014) Parentage assessment of incomplete ossification in larval Japanese flounder by microsatellite DNA markers. Aquaculture 420-1:S98–S103
Sawayama E, Kitamura S-I, Nakayama K, Ohta K, Okamoto H, Ozaki A et al. (2019a) Development of a novel RSIVD-resistant strain of red sea bream (Pagrus major) by marker-assisted selection combined with DNA-based family selection. Aquaculture 506:188–192
Sawayama E, Nakao H, Kobayashi W, Minami T, Takagi M (2019b) Identification and quantification of farmed red sea bream escapees from a large aquaculture area in Japan using microsatellite DNA markers. Aquat Living Resour 32:26
Sawayama E, Noguchi D, Nakayama K, Takagi M (2018) Identification, characterization, and mapping of a novel SNP associated with body color transparency in juvenile red sea bream (Pagrus major). Mar Biotechnol 20:481–489
Sawayama E, Takagi M (2011) Genetic factors associated with transparency of juvenile red sea bream, Pagrus major. Nippon Suisan Gakkaishi 77:630–638
Sawayama E, Takagi M (2012) Genetic investigation of artificially raised red sea bream with abnormal vertebrae formation. Nippon Suisan Gakkaishi 78:62–68
Sawayama E, Takagi M (2016) Genetic diversity and structure of domesticated strains of red sea bream, Pagrus major, inferred from microsatellite DNA markers. Aquac Res 47:379–389
Sawayama E, Tanizawa S, Kitamura SI, Nakayama K, Ohta K, Ozaki A et al. (2017) Identification of quantitative trait loci for resistance to RSIVD in red sea bream (Pagrus major). Mar Biotechnol 19:601–613
Sawayama E, Takagi M (2015) Parental contribution and growth hormone gene polymorphisms associated with growth phenotypes of red sea bream Pagrus major in mass production: a case study. Aquac Rep. 2:144–151
Shin GH, Shin Y, Jung M, Hong JM, Lee S, Subramaniyam S et al. (2018) First draft genome for red sea bream of family Sparidae. Front Genet 9:643
Smith R, Sheppard K, DiPetrillo K, Churchill G (2009) Quantitative trait locus analysis using J/qtl. Methods Mol Biol 573:175–188
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Teletchea F, Fontaine P (2014) Levels of domestication in fish: implications for the sustainable future of aquaculture. Fish Fish 15:181–195
Trijuno DD, Yoseda K, Hirokawa J, Tagawa M, Tanaka M (2002) Effects of thyroxine and thiourea on the metamorphosis of coral trout grouper Plectropomus leopardus. Fish Sci 68:282–289
Trubiroha A, Gillotay P, Giusti N, Gacquer D, Libert F, Lefort A et al. (2018) A rapid CRISPR/Cas-based mutagenesis assay in zebrafish for identification of genes involved in thyroid morphogenesis and function. Sci Rep. 8:5647
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Weber G, Rabbiosi S, Zamproni I, Fugazzola L (2013) Genetic defects of hydrogen peroxide generation in the thyroid gland. J Endocrinol Invest 36:261–266
Yi RH, Zhu WB, Yang LY, Lan L, Chen Y, Zhou JF et al. (2013) A novel dual oxidase maturation factor 2 gene mutation for congenital hypothyroidism. Int J Mol Med 31:467–470
Zamproni I, Grasberger H, Cortinovis F, Vigone MC, Chiumello G, Mora S et al. (2008) Biallelic inactivation of the dual oxidase maturation factor 2 (DUOXA2) gene as a novel cause of congenital hypothyroidism. J Clin Endocrinol Metab 93:605–610
Zheng X, Ma SG, Qiu YL, Guo ML, Shao XJ (2016) A novel c.554+5C>T mutation in the DUOXA2 gene combined with p.R885Q mutation in the DUOX2 gene causing congenital hypothyroidism. J Clin Res Pediatr Endocrinol 8:224–227
Acknowledgements
The authors greatly appreciate the staff of Marua Suisan Co., Ltd. for fish sampling and rearing experiments. This work was supported by JSPS KAKENHI Grant Numbers JP15H00471, JP16H00480, JP17H00469.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Statement
All experiments described in this paper were carried out in accordance with the Guide for the care and use of laboratory animals from Nihon University.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sawayama, E., Handa, Y., Nakano, K. et al. Identification of the causative gene of a transparent phenotype of juvenile red sea bream Pagrus major. Heredity 127, 167–175 (2021). https://doi.org/10.1038/s41437-021-00448-3
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-021-00448-3
