
Abstract
Genomic selection has become a reality in plant breeding programs with the reduction in genotyping costs. Especially in maize breeding programs, it emerges as a promising tool for predicting hybrid performance. The dynamics of a commercial breeding program involve the evaluation of several traits simultaneously in a large set of target environments. Therefore, multi-trait multi-environment (MTME) genomic prediction models can leverage these datasets by exploring the correlation between traits and Genotype-by-Environment (G×E) interaction. Herein, we assess predictive abilities of univariate and multivariate genomic prediction models in a maize breeding program. To this end, we used data from 415 maize hybrids evaluated in 4 years of second season field trials for the traits grain yield, number of ears, and grain moisture. Genotypes of these hybrids were inferred in silico based on their parental inbred lines using single nucleotide polymorphisms (SNPs) markers obtained via genotyping-by-sequencing (GBS). Because genotypic information was available for only 257 hybrids, we used the genomic and pedigree relationship matrices to obtain the H matrix for all 415 hybrids. Our results demonstrated that in the single-environment context the use of multi-trait models was always superior in comparison to their univariate counterparts. Besides that, although MTME models were not particularly successful in predicting hybrid performance in untested years, they improved the ability to predict the performance of hybrids that had not been evaluated in any environment. However, the computational requirements of this kind of model could represent a limitation to its practical implementation and further investigation is necessary.
Similar content being viewed by others


Introduction
The Brazilian maize production is currently concentrated in the second season, from February to June, representing more than 66% of the production in the 2017/2018 season (Conab 2019). The second season is an alternative crop rotation system in the Center-South region, with maize grown mostly after soybean, contributing to a greater profitability of the Brazilian agribusiness. However, this ensuing season poses some challenges, such as greater disease pressure and, especially, water deficiency stress. Due to climate changes and the limitation of water resources, yield stability even under water stress is a highly desirable feature in agriculture nowadays (Cooper et al. 2014). Much of the progress made in growing maize in the second season is due to genetic improvement for drought tolerance.
Maize breeding programs for the second season target genotypes that are highly productive under normal growing conditions, but which are able to maintain good performance even under conditions of water scarcity. The biggest challenge faced by breeders remains in the fact that grain yield is a quantitative trait, strongly influenced by environmental effects and showing low heritability under stress conditions (Comstock 1978; Hallauer and Miranda Filho 2010). Therefore, to increase the experimental precision of phenotypic evaluations under water deficiency, a large number of replicates and adequate plot sizes are required (Edmeades et al. 1999; Bãnziger et al. 2000). However, phenotyping accounts for a large part of the cost of a plant breeding program, limiting progress by restricting the number of evaluated genotypes and the sizes of experiments.
A large number of hybrids can be obtained from the cross of a relatively small number of lines in a maize breeding program (Technow et al. 2014). Due to the financial unfeasibility of evaluating all these possible hybrids in field trials, predicting hybrid performance through genomic selection is an attractive alternative to maize breeders. Since proposed by Meuwissen et al. (2001), genomic selection models have been applied to a variety of crops and became an important tool in maize hybrid breeding (Bernardo 2009; Massman et al. 2013; Dias et al. 2018; Fritsche-Neto et al. 2018; Han et al. 2018). Besides the opportunity to reduce costs and labor involved in field trials, this approach allows an early and more efficient selection, increasing genetic gains. These models were initially proposed and applied in a univariate context, by using a separate model for a single environment and a single-trait. However, breeders commonly evaluate several traits simultaneously in a large set of environments, because elite genotypes should concentrate favorable alleles for various traits of interest and perform well in different target environments. As a prime consequence, the use of univariate approaches might not meet the reality of breeding programs that aim to estimate the magnitude of G×E interaction and explore the genetic correlation between important agronomic traits.
The presence of genetic correlation between quantitative traits implies that measures in one trait indirectly provide information about other traits. From a breeding standpoint, such information can be used to improve the predictive ability of genomic selection, as reported in different species (Marchal et al. 2016; Fernandes et al. 2018; Lyra et al. 2017; Lado et al. 2018). Multivariate genomic selection models, known as multi-trait models, allow the information between secondary traits to be explored through modeling of the covariance between them. The main factors that have been reported to contribute to increasing predictive ability of multi-trait models are: traits highly correlated with the trait of interest and low-heritability coefficients for the target trait, but high for the correlated trait (Calus and Veerkamp 2011; Jia and Jannink 2012; Guo et al. 2014; Dos Santos et al. 2016; Marchal et al. 2016; Lyra et al. 2017; Covarrubias-Pazaran et al. 2018). In maize, grain yield is the trait of major interest and it is a direct function of the following components: number of ears per plant, number of rows of grain in the ear, number of grains per row, ear length, ear diameter, average grain weight, and grain depth (Jugenheimer 1976). Considering that such secondary traits are less complex, present higher heritability coefficients, and, ultimately, are highly correlated with grain yield, they are feasible to perform indirect selection for grain yield. Several studies reported higher predictive abilities using models that consider high-heritability secondary trait information combined to grain yield (Henderson and Quaas 1976; Mrode and Thompson 2005; Malosetti et al. 2008; Piepho et al. 2008).
In addition to the correlation between traits, the G×E interaction is also a relevant issue to plant breeders. Burgueño et al. (2012) were the first to accommodate G×E interaction in the context of genomic selection. Following this study, others also examined the possibility of increasing the predictive ability in several crops by incorporating the G×E interaction (Lopez-Cruz et al. 2015; Cuevas et al. 2016; Ferrão et al. 2017; Sousa et al. 2017; Roorkiwal et al. 2018). Proper understanding of G×E interaction provides valuable information and can help breeders to predict completely untested combinations of hybrids and environments using cross-validation schemes.
The large amount of phenotypic data collected in breeding programs across years is a valuable source of information, of which genomic selection is recently taking advantage. Nonetheless, the quality and unbalanced nature of these historical data raise a new challenge to plant breeders—how to optimally exploit this kind of data (Gapare et al. 2018). Few studies have simultaneously assessed MTME models for genomic selection (Montesinos-López et al. 2016; 2018a, 2019b, 2019c, 2019d; Gomes Torres et al. 2018; Ward et al. 2019). Therefore, our objectives were to: (i) evaluate the applicability of a MTME model in the maize breeding context, (ii) compare the results of using this model with its univariate counterparts, and (iii) predict completely new and untested hybrids and years. To this end, historical data from three traits from second season maize hybrids was used.
Material and methods
Plant material
The genetic material consisted of 415 hybrids evaluated in field trials for four years (2006–2009) in Campo Mourão, Paraná, Brazil. Of the 415 hybrids, 304 are single cross hybrids, 76 are triple cross hybrids, 19 are double cross hybrids, and 16 are commercial checks. The experimental design of the phenotypic trials from 2006 to 2008 was a 10 × 10 squared lattice design with two replicates, where 100 hybrids were evaluated. In the year 2009, 125 hybrids were evaluated side-by-side in two trials. In each trial, 60 hybrids and four common checks were evaluated using an 8 × 8 lattice design with two replicates. The connection across years was based in a few common checks (Table S1).
The evaluated traits were grain yield (GY), determined by weighing all the grains in each plot, adjusted to 13% of grain moisture and converted to tons per hectare (t/ha); number of ears (NE), consisted of counting all ears in each plot; and percentage of grain moisture (GM), assessed with the Wintersteiger Classic Plot Combine automatic harvester (Wintersteiger AG, Mettmach, Austria), which automatically weighs each parcel and infers the moisture via NIRS (near-infrared spectroscopy).
Phenotypic analysis
We computed the best linear unbiased estimation (BLUE) for each trial and trait, using the following mixed model:
where \(y_{{\mathrm{ijk}}}\) is the phenotype of the ith genotype in block j, replicate k; μ is the common intercept; rk is the fixed effect of replicate k; gi is the fixed effect of the ith genotype; bj(k) is the random effect of block j, in replicate k, such that \(b_{\mathrm{j}\left(\mathrm{k}\right)}\sim\mathrm{MVN}\left(0, \boldsymbol{I}\sigma_{b}^{2}\right)\); and εijk is a random non-genetic effect, where \(\varepsilon _{{\mathrm{ijk}}} \sim {\mathrm{MVN}}\left( {0,{\boldsymbol{I}}\sigma _\varepsilon ^2} \right)\); where MVN refers to the multivariate normal distribution and I is an identity matrix. Outliers were removed by deleting observations with residuals that deviated more than four times the standard deviation.
The broad-sense and narrow-sense heritability were computed based on model (1), but considering the genotype effects as random. Also, in order to compute the narrow-sense heritability we assumed that \(g_{\mathrm{i}}\sim {\mathrm{MVN}}\left( {0,\,{\mathbf{H}}\sigma _G^2} \right)\), where H represents the relationship matrix of additive effects. We then estimated the heritability based on the following equation:
where \(\sigma _G^2\) is the total genetic variance component and the additive variance component for the broad-sense and narrow-sense heritability, respectively, and \(\sigma _e^2\) is the residual variance component. The BLUEs and heritability coefficients were estimated using the ASReml-R package version 3.0 (Butler et al. 2009) in the R environment v.3.5.1 (R Development Core Team 2019).
Genotypic data
A collection of 1060 maize inbred lines from the Embrapa Maize and Sorghum breeding program, Brazil was genotyped. Of this total, 228 lines are parents of the hybrids used in this study. We performed DNA extraction from young leaves based on the cetyltrimethylammonium bromide method (Saghai-Maroof et al. 1984). DNA samples were quantified using the Fluorometer Qubit® 2.0, following the manufacturer’s instructions (Life TechnologiesTM, Carlsbad, CA, USA). Samples were also evaluated on 1% agarose gel in Tris-acetate-EDTA buffer, stained with GelRedTM (Biotium, Fremont, CA, USA) and recorded under UV light in the Imager Gel Doc L-PIX (Loccus Biotecnologia, Cotia, SP, Brazil).
Genotyping-by-Sequencing (GBS) was carried out at the Genomic Diversity Facility at Cornell University (Ithaca, NY, USA) using the standard GBS protocol (Elshire et al. 2011) with the ApeKI restriction enzyme. The inbred lines were genotyped in two different batches: first, we genotyped eight libraries of 96 samples each, with one HiSeq 2500 sequencing lane per library; next, we genotyped one library of 384 samples with NextSeq500 in a single lane. Tags were aligned to the B73 reference genome (AGPv3) (Law et al. 2015) using the Bowtie2 aligner (Langmead and Salzberg 2012). Then, SNPs were called using the GBSv2 Discovery Pipeline, available in the software TASSEL v. 5.2.28 (Glaubitz et al. 2014). We applied filters for Minor Allele Frequency (MAF) less than 5%, inbreeding coefficient less than 0.8 and removed indels and non-biallelic markers. Heterozygous loci were treated as missing data. Subsequently, we performed imputation of missing data using Beagle software version 4.1 (Browning and Browning 2016). Because Beagle can introduce heterozygous genotypes, we carried out an additional filtering step by removing heterozygous loci. Finally, from the genotypes of the 228 parental lines we inferred, in silico, the genotypes of 257 single cross hybrids.
H matrix
The hybrids used in this study were originated from 296 inbred lines that belong to three different heterotic groups: Dent (116 lines), Flint (126 lines), and another group herein denominated group C (54 lines). Pedigree information was available for all 415 hybrids, but only 257 of those were (indirectly) genotyped. In this situation, the use of the single-step approach, where the pedigree relationship matrix A and the genomic relationship matrix G are combined into one matrix called H, is a practical way to combine these two sources of information (Legarra et al. 2009; Misztal et al. 2009; Aguilar et al. 2010; Christensen and Lund 2010).
The genomic relationship matrix G was computed following the method described by Yang et al. (2010). The pedigree relationship matrix A was computed based on Henderson’s recursive method described in Mrode and Thompson (2005). We implemented the H matrix using the two scaling factors, τ and ω, as proposed by Misztal et al. (2010) and Tsuruta et al. (2011):
We further evaluated the effect of these factors on the accuracies of genomic prediction models. Using the approach presented by Martini et al. (2018), we searched for the optimal values of τ and ω by evaluating 420 combinations, varying both parameters on grids defined by the intervals [−1, 1] for ω and [0.1, 2] for τ, in steps of size 0.10 in both cases. To evaluate the performance of each parameter combination, we constructed 420 different H matrices, one for each combination of the scaling factors, and used these to estimate the breeding values using the single-step procedure for each single-trait single-environment (STSE) model fitted. The G, A, and H matrices were obtained using the R package AGHmatrix (Amadeu et al. 2016).
Genomic prediction models
The genomic prediction model used in this study was the GBLUP (Genomic Best Linear Unbiased Prediction) (VanRaden 2008) and its multivariate version (Calus and Veerkamp 2011). We fitted the univariate and multivariate models via a Bayesian approach, as detailed below.
Single-trait single-environment model (STSE)
Using the STSE model the genomic estimated breeding values (GEBV) were obtained for each of the three traits evaluated, separately for each of the four environments, as follows:
where yi is the previously obtained BLUE of the ith genotype (i = 1, …, n), where n indicates the number of hybrids evaluated in the environment at hand; μ is the intercept; Gi is the random effect of the ith genotype, such that \(G_i\sim {\mathrm{MVN}}({0,\,{\mathbf{H}}\sigma _g^2})\), \(\sigma _g^2\) is the genetic variance component; and εi is a random non-genetic effect, with εi ~ MVN(0, Iσ2), σ2 is the residual variance component. H represents the relationship matrix of additive effects and I is an identity matrix for the residual effects.
Multi-trait single-environment model (MTSE)
Combining information of the three evaluated traits, separately in each of the four environments, we obtained the GEBV using the multi-trait single-environment (MTSE) model:
where \(y_{ic}\) is the BLUE of the ith genotype for trait c (c = 1, …, 3); μc is the intercept for trait c; Gic is the random effect of the ith genotype for trait c, \(G_{ic}\sim {\mathrm{MVN}}({0,\,\sigma _g^2{\mathbf{H}} \otimes {\mathbf{\Sigma }}_{\mathbf{c}}})\); and εi is a random non-genetic effect, \(\varepsilon_{ic}\sim {\mathrm{MVN}}({0,\,\sigma^2{\mathbf{I}} \otimes {\mathbf{R}}_{\mathbf{c}}})\). In this model, Σc is the variance-covariance (VCOV) matrix for the additive genetic effects of the three traits, with dimension 3 × 3. Rc represents the VCOV matrix for the residual effects of the three traits, also with dimension 3 × 3. We assumed an unstructured form for the genetic Σc and residual Rc VCOV matrices, which allows the assumption of heterogeneity of variance and presence of a specific genetic correlation for each combination of trait and environment. It is important to note that trait main effects are still estimable, despite not being explicitly included in the model formula, for a detailed explanation see Ward et al. (2019) and Isik et al. (2017).
Single-trait multi-environment model (STME)
Through the single-trait multi-environment (STME) model, we obtained the GEBV separately for each of the three traits, but jointly modeling the four environments (years), as follows:
where \(y_{ij}\) is the BLUE of the ith genotype, in the jth environment (j = 1,…,4); μj is the intercept in the jth environment; Gij is the random effect of the ith genotype, in the jth environment, with \(G_{ij}\sim {\mathrm{MVN}}({0,\,\sigma _g^2{\mathbf{H}} \otimes {\mathbf{\Sigma }}_{\mathbf{j}}})\); and εij is a random non-genetic effect, such that \(\varepsilon_{ij}\sim {\mathrm{MVN}}({0,\,\sigma^2{\mathbf{I}} \otimes {\mathbf{R}}_{\mathbf{j}}})\). Σj is the VCOV matrix for the additive genetic effects in the j environments, with dimension 4 × 4. Rj represents the VCOV matrix for the residual effects in the j environments, with dimension 4 × 4. We again assumed an unstructured form for the genetic Σj and residual Rj VCOV matrices and implicitly modeled the environment main effects.
Multi-trait multi-environment model (MTME)
In our most complex model, for MTME genomic selection, we jointly modeled all traits and environments, in order to obtain the GEBV for each trait in each environment:
where \(y_{ijc}\) is the previously obtained BLUE of the ith genotype, in the jth environment, for trait c; μjc is the intercept in the jth environment, for trait c; Gijc is the random effect of the ith genotype, in the jth environment, for the trait c, \(G_{ijc}\sim {\mathrm{MVN}}( {0,\,\sigma _g^2{\mathbf{H}} \otimes {\Sigma}_{{\mathbf{jc}}}} )\); and εijc is a random non-genetic effect, \(\varepsilon_{ijc}\sim {\mathrm{MVN}}({0,\,\sigma^2{\mathbf{I}} \otimes {\mathbf{R}}_{\mathbf{jc}}})\). Σjc represents the VCOV matrix for the additive genetic effects in the four environments for the three traits, with dimension 12 × 12. In this case, this matrix models variances and covariances for all combinations of traits and environments. Similarly, Rjc represents the VCOV matrix for the residual effects in each trait × environment combination, with dimension 12 × 12. We assumed an unstructured form for the genetic and residual VCOV matrices and implicitly modeled the trait and environment main effects.
Cross-validation schemes
To assess the performance of each model we used the predictive ability as measured by cross-validation. To this end, we implemented three different schemes. In our first cross-validation scheme (hereinafter denoted as CVR), the complete pool of individuals was randomly split in five folds, such that four of them were used as a training set, while the remaining group was used as a testing set. This procedure was repeated five times, using a different set of individuals as the testing set each time. Therefore, at the end of the process GEBVs were calculated for all individual. This CVR scheme was applied to models 4, 5, 6, and 7 as described above. In order to compare the models applied to single-environments with those applied to multi-environments, we also evaluated the CVR scheme using the same training/testing partition of single-environment models to the multi-environment ones.
In the multi-environment context, as described above for models 6 and 7, we evaluated two different cross-validation schemes in order to take advantage of the correlated information between environments, according to the ideas presented by Burgueño et al. (2012). First, we aimed to measure the ability of the model to predict the performance of hybrids that had not been evaluated in any environment (hereinafter denoted as CV1). In CV1 we randomly assigned the hybrids to a 5-fold cross-validation scheme, but in this case ensuring that hybrids in the testing set had not been evaluated in any environment. Alternatively, to assess the ability of the model to predict performance based on data from different years we assigned years to folds (such scheme is hereinafter denoted as CV2). In CV2 we had as many folds as years. Therefore, when analyzing the ith fold, hybrids from the ith year were assigned to the testing set and all the hybrids from other years were used as the training set.
In all cross-validation schemes, the predictive abilities were estimated by Pearson’s correlation coefficient between the GEBV and the corresponding BLUE.
Computational implementation
All genomic prediction models were implemented in the MCMCglmm R-package (Hadfield 2010). A total of 30000 MCMC samples were generated, assuming a burn-in period and sampling interval (thin) of 6000 and five iterations, respectively. In particular, for all models we assumed vague priors drawn from an inverse-Wishart distribution. To this end, we set the following hyperparameters: nu = 0.002 and V = diag(n)/n, where n is the number of trait-environment combinations. The use of the so-called non-informative priors ensures that posterior distributions reflect mainly the information from the data and, therefore, that the effect of the prior on the posterior estimate is minimized. We validate this assumption by following guidelines set forth in (Wilson et al. 2010). Specifically, we tested different hyperparameters for the prior distributions, in order to check whether our results were robust regardless of the specified prior.
To check the convergence of the models we used the Geweke criteria (Geweke 1992) implemented in the coda R-package (Plummer et al. 2006), as well as the visual inspection of trace plots of the chains. All code used to implement the genomic prediction models is available at the following repository: https://github.com/amanda-avelar-oliveira/MTME, as well as in the supplementary material (File S1).
Results
H matrix
Based on the evaluation of the 420 combinations of τ and ω scaling factors for each STSE model fitted, we found common optimal values of τ = 0.1 and ω = −0.8. It is important to note that the maximum predictive ability did not substantially differ between the parameter combinations tested; for example, they ranged from 0.33 to 0.42 for GY in 2006.
The heatmap of the H matrix across hybrids grown in different years and common checks showed that the patterns between years differed considerably, with hybrids grown in 2008 being less correlated with the others (Fig. 1). We stress that 2008 was the year with less hybrids (34) for which genotypic information was available (Table S1). Among the 11 checks, nine were exclusive to a single environment, and only four were genotyped (Table S1). We can also observe a pattern of lower relatedness between the checks (Fig. 1).

A total of 415 hybrids were evaluated from 2006 to 2009. Each colored cell represents the relationship between a pair of hybrids.
Genetic parameters
Broad-sense and narrow-sense heritability coefficients varied considerably between traits (Table 1). Overall, the heritabilities were higher for GM and lower for NE, ranging from 0.70 to 0.91 and 0.13 to 0.55, respectively. For GY the broad-sense heritability did not show large variation among years, ranging from 0.51 in 2008 to 0.59 in 2007. However, we observed larger variation between years for the narrow-sense heritability, from 0.18 in 2006 to 0.41 in 2008. The lowest narrow-sense heritability for NE was seen in 2006, with a coefficient of 0.03. The differences between broad- and narrow-sense heritabilities were less pronounced for GM, with no observed difference in 2007 (h2 = 0.91). The phenotypic means also varied considerably between years, ranging from 3.80 to 8.04, 34.02 to 40.19, and 16.43 to 31.88, for GY, NE, and GM, respectively.
Genetic correlations estimated based on the full MTME model showed that correlations varied noticeably across years, for each of the three traits (blocks closer to the diagonal in Fig. 2). For GY, 2009 showed lower correlation coefficients, presenting negative correlations with years 2007 (−0.55) and 2008 (−0.11). Genotype effects in 2006 were negatively correlated with all others for NE. In general, the GM was the trait with lowest correlation between years, particularly for 2006 and 2009. However, the years 2007 and 2008 showed the highest correlation (0.56) compared to the other traits.

Cells indicate the Pearson pairwise correlation coefficient between combinations of traits and years. Colors represent positive (blue) and negative (red) correlations.
Overall, we observed a large number of near zero and negative values of genetic correlation between traits. Also, the correlations between traits varied considerably across years. GY and GM were more positively correlated, with the largest correlation between GY in 2007 and GM in 2008 (0.87). On the other hand, GY and GM presented the largest negative correlation in 2006 (−0.7).
As might be expected, the genetic correlations estimated based on the full STME models were higher than those obtained with the full MTME model (Figs. S1–S3). Overall, GM and GY showed high and positive correlations, except between 2008 and 2009 for GM and between 2007 with 2006 and 2009 for GY. On the other hand, NE presented some high and negative correlations, reaching −0.94 between 2007 and 2009. When comparing the correlations between traits estimated based on the full MTSE models, we observed that the year 2008 presented the lowest correlations (Figs. S4–S7).
Genomic prediction
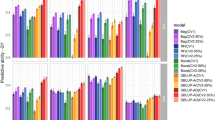
Predictive abilities varied considerably when comparing STSE and MTSE models (Fig. 3). The MTSE models were always superior to the STSE, usually showing substantial differences. Only in 2007, for NE, the use of multivariate model resulted in a slightly superior predictive ability (0.26 vs. 0.32). Within each trait, the predictive abilities differed greatly across years. For example, values for NE ranged from 0.26 in 2007 to 0.48 in 2009 and 0.32 in 2007 to 0.92 in 2009 for the STSE and MTSE models, respectively.

The traits evaluated were grain yield (GY), number of ears (NE) and grain moisture (GM), using single-trait single-environment (STSE) and multi-trait single-environment (MTSE) models. Diamonds correspond to the mean predictive abilities.
For the ME models using the CVR cross-validation scheme (Fig. 4), we observed that the predictive abilities were lower than those observed with the SE models (Fig. 3). Only for GY did the MTME model outperform the STME model in terms of predictive ability, but we note that both values were low (0.07 for STME and 0.15 for MTME). For GM the difference between STME and MTME was low, but the variance was higher in the MTME scenario. On the other hand, NE showed higher variance and predictive ability with the STME model (0.36) compared to the MTME model (0.28). When we used the same training/testing partition of single-environment models to fit multi-environment models, we observed that the multi-trait models outperformed all the others (Fig. 5). However, in some cases the MTME models were inferior to the MTSE. Besides that, specifically for NE in 2006 the MTME model was inferior to all others, including the ST models.

The traits evaluated were grain yield (GY), number of ears (NE) and grain moisture (GM), using single-trait multienvironment(STME) and multi-trait multi-environment (MTME) models. Diamonds correspond to the mean predictive abilities.

The traits evaluated were grain yield (GY), number of ears (NE) and grain moisture (GM), using single-trait single-environment (STSE), single-trait multi-environment (STME), multi-trait single-environment (MTSE) and multi-trait multi-environment (MTME) models. Diamonds correspond to the mean predictive abilities.
The prediction of non-evaluated hybrids using the cross-validation scheme CV1 showed similar predictive abilities for GY when compared to the cross-validation scheme CVR. However, for NE and GM the multi-trait models also outperformed the single-trait ones when using the cross-validation scheme CV1 (Fig. 6).

The traits evaluated were grain yield (GY), number of ears (NE) and grain moisture (GM), using single-trait multi environment (STME) and multi-trait multi-environment (MTME) models. Diamonds correspond to the mean predictive abilities.
Under cross-validation scheme CV2, the prediction abilities were very variable, with lower boundaries spanning-negative-predictive values (Fig. 7). This reflects difficulties in accurately predicting hybrid performance in different years. However, it is important to note that for some specific years and traits the predictive abilities achieved with CV2 were comparable to those obtained with SE models. For example, in 2008 the predictive ability of the STME model for GY was 0.50, similar to the value of 0.36 found with STSE in the same year (Figs. 3 and 7). The comparison between MT and ST models when using the CV2 scheme did not show any clear pattern, with substantial variation across traits and years.

The traits evaluated were grain yield (GY), number of ears (NE) and grain moisture (GM), using single-trait multi-environment (STME) and multi-trait multi-environment (MTME) models.
Discussion
Genomic prediction models have been widely adopted in plant breeding of a variety of species, especially in maize (Bernardo 2009; Massman et al. 2013; Dias et al. 2018; Fritsche-Neto et al. 2018; Han et al. 2018). However, the adoption of models that simultaneously take into account multiple traits and environments—referred here as MTME models—have been limited and only recently has gained attention in the literature (Montesinos-López et al. 2016, 2018a, 2019b, 2019c, 2019d; Gomes Torres et al. 2018; Ward et al. 2019). In this study, we applied genomic prediction to MTME trials of second season maize hybrids and compared their predictive abilities with univariate models.
It is well known that the genetic correlation between traits, as well as the fact that the trait of interest be of low heritability and the correlated trait be of high heritability, are key factors for the success of multi-trait models (Calus and Veerkamp 2011; Jia and Jannink 2012; Guo et al. 2014). However, when major quantitative trait loci (QTL) are not present, that is, for complex polygenic traits, the benefits of multi-trait models are limited even with differences in the heritabilities among highly correlated traits (Jia and Jannink 2012). Our results indicated that multi-trait models can outperform their single-trait counterparts. Similar results were also found in studies based on other maize datasets using genomic prediction models (Montesinos-López et al. 2016, 2019c; Gomes Torres et al. 2018). However, other previous studies reported little benefit of applying multivariate models (Dos Santos et al. 2016; Lyra et al. 2017; Lado et al. 2018). These results can be explained by the low to moderate values of correlation and heritabilities across the traits evaluated, which collectively are expected to hinder multi-trait selection. The challenges faced by breeders under scenarios of low heritability and low genetic correlation among traits are not specific to genomic selection, but inherent to the multi-environment multi-trait breeding process.
Besides the correlation between traits, a model that also accommodates the G×E interaction mimics in a more realistic way the type of data generated in plant breeding programs, where genotypes are evaluated for multiple traits in different environments. Assuming that in single-environment models the training and testing sets are exposed to the same environmental effects, it is biologically reasonable to expect higher predictive abilities than in multiple-environment scenarios. Therefore, when we compared STSE and MTSE approaches, we noticed that the predictive performance of the multivariate models often outperformed the univariate ones. However, when we compared SE models with ME models we observed the opposite behavior. It is worthwhile to highlight that the correlations and heritabilities varied considerably across years, showing the challenges of dealing with the quality and unbalanced nature of our historical data.
In combination, we also assessed the predictive abilities of different cross-validation schemes that mimic real scenarios in the maize breeding program. In the so-called CVR scheme, a complete pool of individuals was randomly split in five folds. For CV1, we randomly assigned the hybrids to a 5-folds scheme, ensuring that hybrids in the testing set had not been evaluated in any environment. Finally, in CV2, we assigned years to folds. For GY, we found similar results when comparing the CVR with CV1 schemes. However, for NE and GM the CV1 scheme showed higher predictive abilities using multi-trait models. Notably, for this particular dataset, the CVR and CV1 schemes are in fact very similar, because few hybrids are common between years. In any case, for two traits we did observe an advantage of multivariate models when hybrids had not been evaluated in any environment, despite this limited connection between trials. The most challenging scenario was the prediction of hybrid performance in different years, as we observed with our cross-validation scheme CV2.
Ward et al. (2019) applied MTME genomic prediction models to unbalanced wheat trials and reported little or no advantage when using the CV1 cross-validation scheme to predict the introduction of new genotypes. However, when genotypes were tested in some environments but not in others, a scenario simulated in the CV2 cross-validation scheme (note that this CV2 scheme is different from the one used in our study), they reported an increase in the predictive ability for low-heritability traits. On the other hand, Jia and Jannink (2012) using simulated datasets found scenarios in which the use of a multi-trait model outperformed the univariate one under a CV1 scheme. This was particularly true for pairs of traits influenced by a few QTLs that present moderate genetic correlation (r = 0.5), one of them being a very low-heritability trait (h2 = 0.1) and the other a moderate-heritability trait (h2 = 0.5).
It is important to note that the lack of genotypic information for all hybrids prompted the use of the single-step procedure, by combining pedigree records and genomic information to estimate a blended relationship matrix—here referred as H. The impact of using an H matrix in genomic prediction has been widely discussed in the animal breeding context (Pszczola et al. 2011; Christensen et al. 2012; Legarra et al. 2014; Martini et al. 2018; Teissier et al. 2019). Conversely, the use of single-step-procedures for genomic prediction is lagging behind in the plant literature. To this end, we estimated the scaling factors τ and ω,—both important parameters to define how the A and G matrixes are combined—as proposed by Misztal et al. (2010) and Tsuruta et al. (2011). In any case, we note that this blending is just one of several possibilities to approach the problem.
The use of Bayesian inference in this study emerged as an alternative to the traditional Restricted Maximum Likelihood (REML) estimation method. We had previously attempted to fit the same models used here with procedures based on REML, but could not achieve convergence. A similar issue was also reported in a study using MTME genomic prediction models in maize (Gomes Torres et al. 2018). Despite reporting the use of REML, Ward et al. (2019) also documented convergence problems for several traits, highlighting the limitation of this technique when using a multivariate approach. As an alternative, we used algorithms based on Markov Chain Monte Carlo (MCMC) methods and implemented in the MCMCglmm R-package. As a limitation, the computational requirements of the Bayesian method presented here may be challenging for practical applications. We evaluated different variance-covariance structures for the genetic and residual terms, and noticed that the computational time required to fit the more complex structure (unstructured) was not different from the simplest one (identity). Because using an unstructured matrix to model (co)variances reflects assumptions that are biologically more realistic, we chose to model the genetic and residual terms using this kind of structure.
The work presented here is an initial investigation of what can be done with MTME models for prediction of hybrid maize, and we believe that the results are promising to justify further research. One important extension would be to incorporate dominance effects in the genomic prediction models, by leveraging the heterosis phenomenon. Several studies including non-additive effects have been conducted and reported the benefits of taking these effects into account (Dos Santos et al. 2016; Resende et al. 2017; Dias et al. 2018). It is important to assess whether favoring more complicated and computationally intensive MTME models is indeed worthwhile.
Finally, we believe that our work also helps to better understand the practical challenges to successfully applying MTME genomic prediction models to a second season maize breeding program. We demonstrated that the use of MTME models can increase predictive ability when compared to univariate ones. However, in some cases we did not observe any improvement, which can at least partly be explained by the low correlation between traits and small heritability differences that we found. Besides that, the low levels of connection between trials in different environments and the necessity of using the single-step procedure highlight the complexity of the historical data we used. We believe that further research is needed to explore ways of dealing with these limitations, which represent the reality of a commercial maize breeding program. Recently, the use of deep learning in multivariate genomic prediction models was investigated, showing promising results (Montesinos-López et al. 2018a, 2018b, 2019a). These studies indicated that the multi-trait deep learning (MTDL) model was very competitive for performing predictions in the context of GS, with the important practical advantage that it requires less computational resources. For this reason, we believe that more research is needed to investigate the reliability of DL applied to multivariate genomic prediction models. Ultimately, this methodology may be added to the data science toolkit of scientists working on breeding programs. Our study additionally suggests that there is room for further work in optimizing multivariate genomic prediction models in Bayesian and frequentist frameworks, allowing the practical application of these complex models.
Data archiving
Data are available in the Dryad Digital Repository: https://doi.org/10.5061/dryad.9w0vt4bc2
References
Aguilar I, Misztal I, Johnson DL, Legarra A, Tsuruta S, Lawlor TJ (2010) Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score 1. J Dairy Sci 93:743–752
Amadeu RR, Cellon C, Olmstead JW, Garcia AAF, Resende MFR, Muñoz PR (2016) AGHmatrix: R package to construct relationship matrices for autotetraploid and diploid species: a Blueberry Example. Plant. Genome 9:1–10
Bãnziger M, Edmeades G, Beck D, Bellon M (2000) Breeding for drought and nitrogen stress tolerance in maize: from theory to pratice. CIMMITY, Mexico
Bernardo R (2009) Genomewide selection for rapid introgression of exotic germplasm in maize. Crop Sci 49:419–425
Browning BL, Browning SR (2016) Genotype imputation with millions of reference samples. Am J Hum Genet 98:116–126
Burgueño J, de los Campos G, Weigel K, Crossa J (2012) Genomic prediction of breeding values when modeling genotype × environment interaction using pedigree and dense molecular markers. Crop Sci 52:707–719
Butler DG, Cullis BR, Gilmour AR, Gogel BJ (2009) ASReml-R Reference Manual. Release 3. Technical Report, Queensland Department of Primary Industries, Brisbane, Queesland, Australia
Calus MPL, Veerkamp RF (2011) Accuracy of multi-trait genomic selection using different methods. Genet Sel Evol 43:1–14
Christensen OF, Lund MS (2010) Genomic prediction when some animals are not genotyped. Genet Sel Evol 42:1–18
Christensen OF, Madsen P, Nielsen B, Ostersen T, Su G (2012) Single-step methods for genomic evaluation in pigs. Animal 6:1565–1571
Comstock RE (1978) Quantitative genetics in maize breeding. In: Walden DB (ed) Maize breeding and genetics. Wiley, New York, p 191–206
Conab (2019) Companhia Nacional de Abastecimento. Séries históricas. Available via http://www.conab.gov.br. Accessed 07 Jan 2019.
Cooper M, Gho C, Leafgren R, Tang T, Messina C (2014) Breeding drought-tolerant maize hybrids for the US corn-belt: discovery to product. J Exp Bot 65:6191–6204
Covarrubias-Pazaran G, Schlautman B, Diaz-Garcia L, Grygleski E, Polashock J, Johnson-Cicalese J et al. (2018) Multivariate GBLUP improves accuracy of genomic selection for yield and fruit weight in biparental populations of Vaccinium macrocarpon Ait. Front Plant Sci 9:1310
Cuevas J, Crossa J, Soberanis V, Perez-Elizalde S, Perez-Rodriguez P, de Los Campos G et al. (2016) Bayesian genomic prediction of Genotype x Environment interaction kernel regression models. G3 Gene Genome Genet 7:41–53
Dias KODG, Gezan SA, Guimarães CT, Nazarian A, da Costa Silva L, Parentoni SN et al. (2018) Improving accuracies of genomic predictions for drought tolerance in maize by joint modeling of additive and dominance effects in multi-environment trials. Heredity 121:24–37
Dos Santos JPR, De Castro Vasconcellos RC, Pires LPM, Balestre M, Von Pinho RG (2016) Inclusion of dominance effects in the multivariate GBLUP model. PLoS ONE 11:1–21
Edmeades GO, Bolaños J, Chapman SC, Lafitte HR, Banziger M (1999) Selection improves drought tolerance in tropical maize populations: I. Gains in biomass, grain yield, harvest index. Crop Sci 39:1306–1315
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES et al. (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:1–10
Fernandes SB, Dias KOG, Ferreira DF, Brown PJ (2018) Efficiency of multi-trait, indirect, and trait-assisted genomic selection for improvement of biomass sorghum. Theor Appl Genet 131:747–755
Ferrão LFV, Ferrão RG, Ferrão MAG, Francisco A, Garcia AAF (2017) A mixed model to multiple harvest-location trials applied to genomic prediction in Coffea canephora. Tree Genet Genomes 13:1–13
Fritsche-Neto R, Akdemir D, Jannink J-L (2018) Accuracy of genomic selection to predict maize single-crosses obtained through different mating designs. Theor Appl Genet 131:1153–1162
Gapare W, Liu S, Conaty W, Zhu Q-H, Gillespie V, Llewellyn D et al. (2018) Historical datasets support genomic selection models for the prediction of cotton fiber quality phenotypes across multiple environments. G3 Gene Genome Genet 8:1721–1732
Geweke J (1992) Evaluating the accuracy of sampling-based approaches to the calculation of posterior moments. In: Bernardo J, Berger J, Dawid A, Smith AF (eds) Bayesian Statistics 4. Clarendon Press, Oxford, UK, pp 625–631
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q et al. (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9:1–11
Gomes Torres L, Rodrigues MC, Lima NL, Freitas T, Trindade H, Fonseca E, Silva F et al. (2018) Multi-trait multi-environment Bayesian model reveals G x E interaction for nitrogen use efficiency components in tropical maize. PLoS ONE 13:1–15
Guo G, Zhao F, Wang Y, Zhang Y, Du L, Su G (2014) Comparison of single-trait and multiple-trait genomic prediction models. BMC Genet 15:1–7
Hadfield JD (2010) MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R package. J Stat Softw 33:1–22.
Hallauer A, Miranda Filho J (2010) Quantitative genetics in maize breeding, 2.ed. Iowa State University Press, Ames
Han S, Miedaner T, Utz FU, Schipprack W, Schrag TA, Melchinger AE (2018) Genomic prediction and GWAS of Gibberella ear rot resistance traits in dent and flint lines of a public maize breeding program. Euphytica 214:1–20
Henderson CR, Quaas RL (1976) Multiple trait evaluation using relatives records. J Anim Sci 43:1188–1197
Isik F, Holland J, Maltecca C (2017) Genetic Data Analysis for Plant and Animal Breeding, 1st edn. Springer International Publishing, Cham
Jia Y, Jannink JL (2012) Multiple-trait genomic selection methods increase genetic value prediction accuracy. Genetics 192:1513–1522
Jugenheimer RW (1976) Corn improvement, seed production and uses. Wiley-Interscience, New York
Lado B, Vázquez D, Quincke M, Silva P, Aguilar I, Gutiérrez L (2018) Resource allocation optimization with multi-trait genomic prediction for bread wheat (Triticum aestivum L.) baking quality. Theor Appl Genet 131:2719–2731
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Law M, Childs KL, Campbell MS, Stein JC, Olson AJ, Holt C et al. (2015) Automated update, revision, and quality control of the Maize genome annotations using MAKER-P improves the B73 RefGen_v3 gene models and identifies new genes. Plant Physiol 167:25–39
Legarra A, Aguilar I, Misztal I (2009) A relationship matrix including full pedigree and genomic information. J Dairy Sci 92:4656–4663
Legarra A, Christensen OF, Aguilar I, Misztal I (2014) Single Step, a general approach for genomic selection. Livest Sci 166:54–65
Lopez-Cruz M, Crossa J, Bonnett D, Dreisigacker S, Poland J, Jannink J-L et al. (2015) Increased prediction accuracy in wheat breeding trials using a marker × environment interaction genomic selection model. G3 Gene Genome Genet 5:569–82
Lyra DH, Mendonça L, de F, Galli G, Alves FC, Granato ÍSC, Fritsche-Neto R (2017) Multi-trait genomic prediction for nitrogen response indices in tropical maize hybrids. Mol Breed 37:1–14
Malosetti M, Ribaut JM, Vargas M, Crossa J, Van Eeuwijk FA (2008) A multi-trait multi-environment QTL mixed model with an application to drought and nitrogen stress trials in maize (Zea mays L.). Euphytica 161:241–257
Marchal A, Legarra A, Sébastien T, Catherine, Carasco-Lacombe Aurore M, Edyana S, Alphonse O et al. (2016) Multivariate genomic model improves analysis of oil palm (Elaeis guineensis Jacq.) progeny tests. Mol Breed 36:1–13
Martini JWR, Schrauf MF, Garcia-Baccino CA, G Pimentel EC, Munilla S, Rogberg-Muñoz A et al. (2018) The effect of the H −1 scaling factors τ and ω on the structure of H in the single-step procedure. Genet Sel Evol 50:1–9
Massman JM, Jung HJG, Bernardo R (2013) Genomewide selection versus marker-assisted recurrent selection to improve grain yield and stover-quality traits for cellulosic ethanol in maize. Crop Sci 53:58–66
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Misztal I, Aguilar I, Legarra A, Lawlor TJ (2010) Choice of parameters for single-step genomic evaluation for type I. In: Proceedings of the 61st annual meeting of the European association for animal production, Heraklion, Vol. 16, p 23–27
Misztal I, Legarra A, Aguilar I (2009) Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information. J Dairy Sci 92:4648–4655
Montesinos-López OA, Montesinos-López A, Crossa J, Toledo FH, Pérez-Hernández O, Eskridge KM et al. (2016) A genomic bayesian multi-trait and multi-environment model. G3 Gene Genome Genet 6:2725–2744
Montesinos-López OA, Montesinos-López A, Crossa J, Gianola D, Hernández-Suárez CM, Martín-Vallejo J (2018a) Multi-trait, multi-environment deep learning modeling for genomic-enabled prediction of plant traits. G3 Gene Genome Genet 8:3829–3840
Montesinos-López A, Montesinos-López OA, Gianola D, Crossa J, Hernández-Suárez CM (2018b) Multi-environment genomic prediction of plant traits using deep learners with dense architecture. G3 Gene Genome Genet 8:3813–3828
Montesinos-López OA, Martín-Vallejo J, Crossa J, Gianola D, Hernández-Suárez CM, Montesinos-López A et al. (2019a) New deep learning genomic-based prediction model for multiple traits with binary, ordinal, and continuous phenotypes. G3 Gene Genome Genet 9:1545–1556
Montesinos-López OA, Montesinos-López A, Crossa J, Cuevas J, Montesinos-López JC, Gutiérrez ZS et al. (2019b) A Bayesian Genomic multi-output regressor stacking model for predicting multi-trait multi-environment plant breeding data. G3 Gene Genome Genet 9:3381–3393
Montesinos-López OA, Montesinos-López A, Hernández MV, Ortiz-Monasterio I, Pérez-Rodríguez P, Burgueño J et al. (2019c) Multivariate bayesian analysis of on-farm trials with multiple-trait and multiple-environment data. Agron J III:1–12
Montesinos-López OA, Montesinos-López A, Luna-Vázquez FJ, Toledo FH, Pérez-Rodríguez P, Lillemo M et al. (2019d) An R package for bayesian analysis of multi-environment and multi-trait multi-environment data for genome-based prediction. G3 Gene Genome Genet 9:1355–1369
Mrode RA, Thompson R (2005) Linear models for the prediction of animal breeding values, 3rd edn. CABI, Boston
Piepho HP, Möhring J, Melchinger AE, Büchse A (2008) BLUP for phenotypic selection in plant breeding and variety testing. Euphytica 161:209–228
Plummer M, Best N, Cowles K, Vines K (2006) CODA: Convergence diagnosis and output analysis for MCMC. R N 6:7–11
Pszczola M, Mulder HA, Calus MPL (2011) Effect of enlarging the reference population with (un)genotyped animals on the accuracy of genomic selection in dairy cattle. J Dairy Sci 94:431–441
R Development Core Team (2019) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.Rproject.org/
Resende RT, Resende MDV, Silva FF, Azevedo CF, Takahashi EK, Silva-Junior OB et al. (2017) Assessing the expected response to genomic selection of individuals and families in Eucalyptus breeding with an additive-dominant model. Heredity 119:245–255
Roorkiwal M, Jarquin D, Singh MK, Gaur PM, Bharadwaj C, Rathore A et al. (2018) Genomic-enabled prediction models using multi-environment trials to estimate the effect of genotype × environment interaction on prediction accuracy in chickpea. Sci Rep. 8:1–11
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Population biology ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Sousa MB, Cuevas J, Couto EG, de O, Pérez-Rodríguez P, Jarquín D, Fritsche-Neto R et al. (2017) Genomic-enabled prediction in maize using kernel models with genotype × environment interaction. G3 Gene Genome Genet 7:1995–2014
Technow F, Schrag TA, Schipprack W, Bauer E, Simianer H, Melchinger AE (2014) Genome properties and prospects of genomic prediction of hybrid performance in a breeding program of maize. Genetics 197:1343–1355
Teissier M, Larroque H, Robert-Granie C (2019) Accuracy of genomic evaluation with weighted single-step genomic best linear unbiased prediction for milk production traits, udder type traits, and somatic cell scores in French dairy goats. J Dairy Sci 102:3142–3154
Tsuruta S, Misztal I, Aguilar I, Lawlor TJ (2011) Multiple-trait genomic evaluation of linear type traits using genomic and phenotypic data in US Holsteins. J Dairy Sci 94:4198–4204
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Ward BP, Brown-Guedira G, Tyagi P, Kolb FL, Van Sanford DA, Sneller CH et al. (2019) Multienvironment and multitrait genomic selection models in unbalanced early-generation wheat yield trials. Crop Sci 59:491–507
Wilson AJ, Réale D, Clements MN, Morrissey MM, Postma E, Walling CA et al. (2010) An ecologist’s guide to the animal model. J Anim Ecol 79:13–26
Yang J, Benyamin B, Mcevoy BP, Gordon S, Henders AK, Nyholt DR et al. (2010) Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42:565–569
Acknowledgements
This research was supported by FAPEMIG (Fundação de Amparo à Pesquisa de Minas Gerais), CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, program PREMIO 2045/2014, grant 23038.007195/2012-39), and Embrapa (Brazilian Agricultural Research Corporation). This study was also financed in part by CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil - Finance Code 001). AAO received a fellowship from CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, PDSE - 88881.187106/2018-01) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
de Oliveira, A.A., Resende, M.F.R., Ferrão, L.F.V. et al. Genomic prediction applied to multiple traits and environments in second season maize hybrids. Heredity 125, 60–72 (2020). https://doi.org/10.1038/s41437-020-0321-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-0321-0
This article is cited by
-
Robotized indoor phenotyping allows genomic prediction of adaptive traits in the field
Nature Communications (2023)
