
Abstract
Understanding how species have responded to past climate change may help refine projections of how species and biotic communities will respond to future change. Here, we integrate estimates of genome-wide genetic variation with demographic and niche modeling to investigate the historical biogeography of an important ecological engineer: the dusky-footed woodrat, Neotoma fuscipes. We use RADseq to generate a genome-wide dataset for 71 individuals from across the geographic distribution of the species in California. We estimate population structure using several model-based methods and infer the demographic history of regional populations using a site frequency spectrum-based approach. Additionally, we use ecological niche modeling to infer current and past (Last Glacial Maximum) environmental suitability across the species’ distribution. Finally, we estimate the directionality and possible spatial origins of regional population expansions. Our analyses indicate this species is subdivided into three regionally distinct populations, with the deepest divergence occurring ~1.7 million years ago across the modern-day San Francisco-Bay Delta region; a common biogeographic barrier for the flora and fauna of California. Our models of environmental suitability through time coincide with our estimates of population expansion, with relative long-term stability in the southern portion of the range, and more recent expansion into the northern end of the range. Our study illustrates how the integration of genome-wide data with spatial and demographic modeling can reveal the timing and spatial extent of historic events that determine patterns of biotic diversity and may help predict biotic response to future change.
Similar content being viewed by others

Introduction
The glacial–interglacial cycles of the Quaternary have long been considered primary drivers of contemporary patterns of biodiversity (Hewitt 2000; Peterson 2009). The dynamic climatic oscillations of this time period were characterized by cooler glacials followed by warmer interglacials, and very short transitions from cool to warm periods (Dansgaard et al. 1993). For example, in North America during the Last Glacial Maximum (LGM) 22,000–18,000 years before present, large ice sheets covered most of the northern portion of the continent, and climatic conditions were generally cooler and drier than at present (Hopkins et al. 2013), except in the southwest where it was wetter (Clark et al. 2012). These global climatic oscillations and associated landscape changes likely influenced the current distribution of biodiversity through repeated reduction, isolation, and subsequent expansion of species across large geographic areas (Hewitt 2004; Svenning and Skov 2004).
Paleoecological, distribution modeling, and genetic evidence support the dynamic changes that species experienced with past climate change. During the Quaternary, many plant and animal species retreated into areas of glacial climate refugia (termed refugia hereafter)—local areas with the appropriate environmental conditions to allow species to persist—and expanded their ranges when environmental conditions were favorable once again (Jackson and Overpeck 2000; Hewitt 2004; Nogués-Bravo et al. 2008). This pattern of range shift is seen in North America during the most recent glacial–interglacial transition from the LGM to the present (Graham et al. 1996; Jackson and Overpeck 2000; Waltari et al. 2007). In addition to range shifts, many species in North America experienced a reduction in habitat during the glacial periods (e.g., Knowles and Massatti 2017; Reid et al. 2018) due to the spread of continental ice sheets during the LGM and the reduction or fragmentation of species preferred habitat (Hewitt 2004; Nogués-Bravo et al. 2008). These dramatic changes to species ranges can leave distinct patterns on the present-day genetic structure of a species (Waltari et al. 2007; Knowles and Massatti 2017; Reid et al. 2019). For example, populations that recolonized areas that were previously covered by glaciers or otherwise inhospitable should show signs of recent rapid genetic expansion (Lessa et al. 2003; Burbrink et al. 2016).
California offers a unique opportunity to study how climate oscillations influence patterns of diversity and differentiation in detail across a wide breadth of ecologically distinct taxa. The California Floristic Province is a recognized diversity hotspot due to the combination of high species endemism and significant conservation threat (Myers et al. 2000). The high topographic complexity of California leads to significant environmental gradients in temperature, precipitation, and many other variables, across latitudes, meridians, and elevations that contribute to the generation and maintenance of genetic and biotic diversity (Parisi 2003; Davis et al. 2008). High topographic complexity has magnified the ecological dynamics and climatic oscillations of the past 3–5 million years (Badgley et al. 2017) leading to repeated broad-scale contractions and expansions of species ranges resulting, in many cases, in concordant patterns of diversity and differentiation across multiple species and taxonomic groups (LaPointe and Rissler 2005; Rissler et al. 2006). In particular, concordant phylogeographic breaks exist in many plant and animal taxa across the San Francisco Bay-Delta, the Monterey Bay, between the northern Sierra Nevada and southern Cascade Mountains, along the western slope of the Sierra Nevada, and across the Tehachapi-Transverse Ranges (reviewed in Schierenbeck 2014). These concordant phylogeographic patterns within California further support the role of repeated climatic oscillations across the geographic landscape as drivers of differentiation and diversity across species (Lessa et al. 2003; Grivet et al. 2006).
Species that should be of particular interest for reconstructing phylogeographic history are those that have numerous ecological connections to their communities. Such ecological connections include predator–prey and host–pathogen relationships, as well as species that are environmental engineers that determine the presence of other community members (i.e. ‘strong interactors’; Soulé et al. 2005). One such species in California is the dusky-footed woodrat (Neotoma fuscipes (Goldman 1910; Hooper 1938; Matocq 2002a, b)). This taxon is well known for the large stick houses (commonly ~1 m in height) that individuals build (Carraway and Verts 1991). These structures provide a stable microclimate in comparison to the outside environment (Brown 1968) not only for the woodrat that occupies the house, but also for the many other small vertebrate and invertebrate species that occupy these structures (Whitford and Steinberger 2010). In addition to their role as environmental engineers, woodrats are the most common prey item of the northern spotted owl (Sakai and Noon 1993; Thome et al. 1999), and one of the primary mammalian hosts of Borrelia burdorferi, the causative agent of Lyme disease (Lane and Brown 1991) and other tick-borne pathogens (Foley et al. 2016). Beyond their central role in the communities they occupy, species of the genus Neotoma are also well known for the rich paleorecord they have left behind. The contents of their complex, multi-chambered houses (e.g. plant fragments, scat, raptor pellets) often become fossilized and have provided a uniquely detailed view of ecological and environmental change through time in western North America (Jackson et al. 2005). These “middens” can persist for thousands of years and have been used to document change in body size (Smith et al. 1995) and community composition across tens of thousands of years (Blois et al. 2010). By understanding the evolutionary history and historical demography of woodrats, we can begin to shed light on species-level dynamics that may have had community-level consequences, and that can be uniquely tested given the existence of the high-resolution paleorecord generated by these animals.
The range of Neotoma fuscipes (sensu Matocq 2002a) extends from western Oregon through the Coast Ranges of northern California, the San Francisco Bay Area and south through the Inner Coast Range, and throughout the northern Sierra Nevada foothills, Modoc Plateau, and Cascade Range (Fig. 1). Using mitochondrial data, Matocq (2002b) showed that N. fuscipes comprises two lineages separated by the San Francisco Bay, and suggested that N. fuscipes had recently expanded into northern California, given relatively low mtDNA diversity across that region. Here, we revisit many of these original collections and substantially augment them with newly available samples, especially from northeastern California (courtesy of the Museum of Vertebrate Zoology and the Grinnell Resurvey Project). In addition to augmented spatial coverage, we also augment our genomic sampling by using high-throughput sequencing across the nuclear genome. We integrate these genetic data with both niche and demographic modeling to provide a temporally comprehensive and spatially detailed view of the demographic and biogeographic history of this species across its distribution. Specifically, we ask (1) does N. fuscipes exhibit significant subdivision within its distribution that coincides with known phylogeographic breaks in other species? (2) Did N. fuscipes experience range contractions/expansions in the recent past despite occurring in largely unglaciated areas, and if so, were there multiple refugial areas? (3) What is the timing of major events in the history of this species including divergence, contraction, and expansion? By integrating genome-wide estimates of genetic variation with niche and demographic modeling, we provide a temporally and spatially detailed view of the history of this taxon across its range.

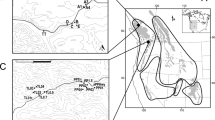
Maximum likelihood tree, Admixture, and Structure results showing three distinct clusters (K = 3): one southern and two northern populations. Colors on the branch tips correspond to the Admixture/Structure population individuals were placed within, and the colored circles on map indicate the proportion of ancestry each individual has to a population. The black circles at nodes indicate bootstrap support >70%.
Materials and methods
ddRAD library preparation and sequencing
DNA was extracted from 71 individual Neotoma fuscipes and 8 N. macrotis (to provide outgroup rooting; Fig. 1; Supplementary Table 1) using the Qiagen DNeasy Blood and Tissue kit (Qiagen, Inc.). All samples used in this study are housed in the Museum of Vertebrate Zoology, University of California, Berkeley. We generated multilocus SNP datasets following the double-digest RADSeq protocol (Peterson et al. 2012). Briefly, we used 500 ng of genomic DNA per sample and digested them overnight with the common-cutting restriction enzyme EcoR1 and the rare-cutting enzyme MSP1. We cleaned the digested samples with Ampure (Invitrogen) beads and eluted the samples with water. These products were tagged with a unique five base-pair (bp) barcode adaptor. We pooled and purified the tagged samples (8–10 individuals per pool) and used a pippin prep to size select for fragments between 300 and 500 bp long. Each library was amplified via PCR with a second index primer to differentiate individuals. Finally, the libraries were sequenced at the U.C. Berkeley QB3 Vincent J. Coates Genomics Sequencing Laboratory on two lanes of an Illumina HiSeq 2500 to produce 100 bp single end reads.
The Illumina HiSeq raw reads were cleaned using the Stacks pipeline (v. 1.21; Catchen et al. 2013). The process_radtags script (from Stacks) was used to filter out low-quality reads and demultiplex the reads according to their unique barcodes. The demultiplexed reads were imported into ipyrad (v. 0.7.17; https://github.com/dereneaton/ipyrad/blob/master/docs/index.rst) pipeline to cluster first within and then among individuals and de novo assemble the reads. We ran several iterations of ipyrad because with next generation sequencing, there can be large amounts of variation between loci due to missing data (Huang and Knowles 2016). We ran ipyrad with all samples (both N. fuscipes and N. macrotis) to include the outgroup for phylogenetic analyses. We then re-clustered the N. fuscipes samples alone to be used in the population structure and demographic analyses focused solely on this taxon. We used two different thresholds for the minimum number of individuals needed to retain a locus to determine the effects of missing data: 50% and 85%, referred to hereafter as the 50% and 85% datasets, respectively (see Appendix S1 for program inputs). This first filtering step controls for the amount of missing data across individuals. Following read clustering, we used the --missing-indv tool in vcftools (v. 0.1.17; Danecek et al. 2011) to calculate the amount of missing data within each individual and removed three samples that had more than 70% missing data. Once we explored the effect of missing data and determined it has little influence on inference of population structure, we ran the range expansion and genetic diversity analyses using a 67% threshold to strike a balance between missing data from natural sequencing variation and the different number of individuals per population. We subsequently re-ran ipyrad on each of the populations identified in the population structure analyses using a 67% threshold.
Phylogenetic analyses
We inferred phylogenetic relationships among sequenced individuals with N. macrotis as the outgroup using RAxML (v. 8.2.12; Stamatakis and Ludwig 2005; Stamatakis 2014) a software program used for large datasets that employs a maximum likelihood (ML) algorithm. Note, this analysis is not appropriate for many phylogenetic questions involving intraspecific SNP data; however, we used this method to understand how individuals grouped together, rather than to infer their specific phylogenetic relationships (following Harrington et al. 2017). To understand the effects of missing data, we ran the RAxML analysis on both the 50% and 85% datasets. Further, we used the full concatenated sequence (invariant and variant sites) to avoid acquisition bias (Leaché et al. 2015). We used rapid bootstrapping (with an automatic stop under the autoMRE criterion), and searched for the best tree under the GTRGAMMA model with final optimization of trees using GTR+Γ. We used Splitstree (V 4.14.8; Huson and Bryant 2006) with only the N. fuscipes individuals to generate a phylogenetic network. We calculated distances between all individuals using the Jukes–Cantor model (Jukes and Cantor 1969) and used the neighbornet method to build the network. Finally, we used these pairwise distance estimates to compare genetic variation within and among clades.
Population structure and isolation by distance
To investigate population structure, we used two model-based and one non-model-based approaches. All methods attempt to determine the optimal number of populations present within the dataset. We used only one SNP per locus for each population structure analysis. We ran Structure, a Bayesian-clustering algorithm that identifies population structure (Pritchard et al. 2000), using 50,000 burn-in generations, 1,000,000 generations, K = 1–5 populations, using the admixture and allele frequencies correlated models, and for 10 iterations. We used the Evanno method (Evanno et al. 2005) in Structure Harvester (v. 0.6.94; Earl and vonHoldt 2012) to determine the optimal number of populations. Because Structure Harvester may not accurately infer the correct number of groups, typically inferring the upper most level of genetic structure (often K = 2; Janes et al. 2017), we used a hierarchical approach, and performed a second set of Structure analyses on each of the initial populations identified (Pritchard et al. 2000). We used 50,000 burn-in generations, 200,000 generations, K = 1–5 populations, using the admixture and allele frequencies correlated models, and for five iterations. We also used Admixture (v. 1.3), a maximum-likelihood approach to determine the optimal number of populations (Alexander et al. 2009). We used cross-validation error values across different values of K to determine the optimal number of populations. Finally, we used the non-model-based method discriminate analysis of principal components (DAPC) in the adegenet package (Jombart 2008; Jombart and Ahmed 2011) in R (v. 3.5.1; R core team 2106) to infer structure. For the DAPC analysis, we used Bayesian information criterion to determine the optimal number of populations. Once optimal populations were identified from each method, we estimated population differentiation by calculating pairwise FST using vcftools (v. 0.1.17; Danecek et al. 2011). To understand the effects of missing data, we ran all of these analyses on the 50% and 85% datasets.
We assessed whether there was a significant signal of isolation by distance (IBD) within the entire species and each regional group identified by Structure. A pattern of IBD is expected to arise at a regional scale once populations reach an equilibrium between dispersal among populations and genetic drift within populations (as shown in Hutchison and Templeton 1999 [Case1]; van Strien et al. 2015). This specific pattern of IBD, showing increased genetic differences with an increase in geographic distance across the whole range, is generally expected to arise in regions that have been stably occupied (Hutchison and Templeton 1999). We plotted the relationship between genetic (FST) and geographic distance among populations within regional genetic groups and determined the significance of these relationships using a Mantel test and 10,000 permutations in the adegenet package (Jombart 2008; Jombart and Ahmed 2011).
Demographic model testing
We used a coalescent-based approach to model the demographic history of Neotoma fuscipes in a temporal framework, specifically with respect to isolation and migration. We selected and parameterized the best-fit demographic model using fastsimcoal2 (FSC2; v2.6.0.3; Excoffier et al. 2013), which estimates demography from the site frequency spectrum (SFS). We followed guidance from Hotaling et al. (2018) in setting up several of our input files. FSC2 allows users to generate hypotheses with differing levels of complexity and uses simulations to estimate the likelihood of competing hypotheses with different parameters. Individuals were assigned using a three-population model based on the population structure analyses (see above), with admixed individuals assigned to the majority population. We generated the observed joint SFS using code developed by Isaac Overcast (https://github.com/isaacovercast/easySFS).
We tested 17 different demographic scenarios (Fig. 2). Briefly, they describe variations of a three-population model that have two divergence events with every possible topology and vary in the amounts of migration and timing in divergence between populations, while also estimating the effective population size (NE) of every regional grouping. Additionally, we modeled trifurcation scenarios with different levels of historical and recent migration events. For Neotoma fuscipes, we used a nuclear mutation rate of 2.5 × 10−8 mutations/site/generation based on estimates from human loci (Nachman and Crowell 2000) and used one-year estimated generation time. For each model we ran 75 replicate FSC2 analyses, each using 250,000 coalescent simulations. We selected the best fit model by calculating AIC and ΔAIC scores to account for number of parameters following the guideline of Excoffier et al. (2013). Once the best model was obtained, we re-estimated parameter values by simulating 100 SFS from the maxL.par file to obtain a mean parameter estimate and 95% confidence intervals. We accounted for N. fuscipes being diploid by dividing the FSC2 estimates in half. For migration rates, we multiplied the values by population size, and then divided by two to account for ploidy.

The parameters included contemporary population size for the three populations, their ancestral population sizes, migration rates between the populations, and the timing of divergence.
Ecological niche models
We generated ecological niche models (EMNs) to infer past areas of refugia. For ENMs, we limited the scope of the analyses to only individuals for which we had genetic data (Supplementary Table 1). Because of the deep divergence between the two northern and the southern populations we modeled each separately (combined north, south). To remove spatial biases, we spatially filtered the dataset to ensure no two localities were within 5 km of one another (Boria et al. 2014) using the R package spThin (Aiello-Lammens et al. 2015). For environmental data, we used Community Climate Simulation Model 3 (CCSM3; Liu et al. 2009). CCSM3 variables are downscaled to 50 × 50 km degree grid cells for North America from 21,000 years ago to present day at 500-year intervals (Lorenz et al. 2016). To approximate modeling assumptions regarding dispersal and biotic interactions more closely, we delimited a custom study region, specifically by drawing a minimum convex polygon around the localities and adding a 3.0° buffer (Anderson and Raza 2010; Barve et al. 2011). We used a machine learning algorithm, maxent (V3.4.1; Phillips et al. 2017, 2006) to infer the ENMs. We calibrated and evaluated the models using a jackknife approach (Pearson et al. 2007) in the ENMeval package in R (Muscarella et al. 2014). To select species-specific model settings approximating optimal levels of complexity, we tuned model settings by varying different combinations of feature class and regularization multiplier (RM; Shcheglovitova and Anderson 2013). To identify the optimal parameter settings, we evaluated model performance using sequential criteria, lowest average omission rate and secondarily on the highest average AUC value (minimizing overfitting and then maximizing discriminatory ability; Shcheglovitova and Anderson 2013; Muscarella et al. 2014). We used the optimal settings to project the ENM for each regional population into current climatic conditions and the LGM.
Range expansion
We inferred origins of recent range expansion within the southern and combined northern populations using the rangeExpansion package in R (Peter and Slatkin 2013). Briefly, this method detects range expansion and gives an estimated location of the origin of expansion. It does so by calculating a directionality index (Ψ), using the genetic data and spatial coordinates, based on allele frequency clines found between multiple populations (Peter and Slatkin 2013). Populations at the expanding edge will tend to have lower genetic diversity because of serial founder events and higher fixation rates due to small effective population sizes (Peter and Slatkin 2015). For each population, we only used loci that were present in at least two-thirds of the individuals.
Genetic diversity
We estimated the population genetic diversity parameter, θ, using ThetaMeta (Adams et al. 2018). This program uses an infinite sites likelihood model to estimate a posterior probability distribution of θ for a given dataset. To understand the population dynamics further, we used the population structure results and calculated θ for each of the three identified populations. For each population, we only used loci that were present in at least two-thirds of the individuals. We used the ThetaMater.M1 simulation to generate a posterior probability distribution of θ with a burnin of 100,000 and a MCMC run of one million generations.
Results
ddRAD data
We obtained 216,373,426 raw single end reads from the Illumina HiSeq run across all individuals. There were 140,817 loci present before ipyrad quality filtering. After filtering, there were 15,334 loci (mean across individuals = 13,576) for the 85% missing-data threshold and 57,860 loci (mean = 43,670) for the 50% missing-data threshold. For the final 50% and 85% datasets, there were 34,434 (85%) and 122,636 (50%) total SNPs, respectively.
Phylogenetic analyses
We obtained very similar tree topologies with RAxML with both the 50% and 85% datasets (Fig. 1; Supplementary Fig. 1A). Essentially, we recovered one monophyletic group of individuals south of the San Francisco Bay-Delta region, and one large monophyletic group of individuals north of the Bay-Delta region (Fig. 1). The northern group is further split into two monophyletic groups, one that comprises the majority of the northern range (North A hereafter), and a second that is primarily restricted to the western slopes of the Sierra Nevada (North B hereafter). The Splitstree results were very similar, clearly separating the northern and southern populations (Supplementary Fig. 2), as well as separating the North A and North B populations.
Inter-individual genetic distances were higher among individuals in the southern clade than in either the North A or North B clades. On average, southern individuals were differentiated from one another by a genetic distance of 0.073 (min.−max.: 0.044–0.097), North A had average pairwise divergence of 0.04 (0.015–0.054) and North B had 0.033 (0.013–0.042; Supplementary Table 2). These distinctions are visually evident (Fig. 1) in the deeper genetic structure of the southern clade in contrast to the shallow, minimal pairwise divergences that characterize both northern clades.
Population structure and isolation by distance
Structure initially gave a K = 2 model as the best fit for the data; however, when we ran Structure on the two subpopulations, the northern population was split further into two distinct populations (Fig. 1) and for the southern population the optimal number of subpopulations was K = 4, though the number of individuals sampled was low in each subpopulation (Supplementary Fig. 3). Although K = 2 was the optimal model in Structure, K = 3 inferred the same northern split of samples we found by running the northern region alone (Fig. 1). Admixture indicated K = 3 as the optimal number of populations based on cross-validation (Fig. 1; Supplementary Tables 3 and 4) regardless of the amount of missing data included. DAPC analysis identified K = 3 as the optimal number of populations for both datasets (Supplementary Figs. 4 and 1C). Both Admixture and Structure identified some admixture between the two Northern populations. All methods assigned the same individuals to the same population across analyses (and datasets), and Admixture and Structure identified the same individuals in the northern group as being admixed. Pairwise FST showed strong differentiation between the southern and northern groups (ave. FST = 0.36) and moderate differentiation between the North A and North B groups (FST = 0.08; Supplementary Fig. 1D; Supplementary Table 5). We adjusted downstream analyses to reflect the population structure results, generally relying on K = 3 except for the niche modeling and range expansion analyses.
The Mantel test showed significant isolation by distance for the overall dataset (r = 0.605, p < 0.001). However, we did not recover a distinct cline, indicating lack of a clear IBD signature (Supplementary Fig. 5; Supplementary Table 6). Each of the regional groups showed a significant relationship between genetic and geographic distance, all following a similar pattern of increasing genetic differentiation with increased geographic distance (Case 1 of Hutchison and Templeton 1999; Fig. 3). The southern region showed a pronounced pattern of IBD (r = 0.731, p < 0.001) and the North A group had a slight pattern of IBD (r = 0.537, p < 0.001). The North B region showed an IBD pattern (r = 0.544, p < 0.001) though the samples from this region occurred over a very small spatial scale.

A North A (r = 0.537, p < 0.001); B North B (r = 0.544, p < 0.001); C South (r = 0.731, p < 0.001). Note that there is significant isolation by distance found within each population.
Demographic modeling
Using FSC2, the model that best fit the data according to AIC was scenario 11 (Table 1; Supplementary Table 7; Fig. 4). This consisted of the two northern populations coalescing most recently, with an older coalescent event between the north and south clusters and both historical and recent admixture (Fig. 4; Supplementary Table 8; see Supplementary Table 9 for confidence intervals). No other scenario had a reasonably good fit (Table 1; Supplementary Table 7).

Note these are the re-estimated parametervalues from 100 simulated Site Frequency Spectrums.
Scenario 11 estimated the two northern populations coalesced about 76,000 ka (73,063–79,644) and the ancestral northern and southern lineages coalesced ~1.72 million years ago (1.71–1.73; Fig. 4). The southern population has the largest effective population size (over 800,000 individuals), North A population has the second largest population size (over 300,000), and North B has the smallest effective population size (~4000). The migration rates between most of the populations (historical and recent) were all less than one individual per year (Fig. 4).
Ecological niche models
The optimal Maxent settings for the northern population was the Hinge feature with a RM = 1.0 (Supplementary Table 10). This model had an AUC of 0.83 and an omission rate of 0.045. The optimal model for the southern population used the Linear and Quadratic features and a RM = 1.5 and had an AUC of 0.88 and an omission rate of 0.091. ENMs for the northern population inferred suitable environment present throughout the entire known range of N. fuscipes for current conditions, and a severe reduction of habitat at the LGM relative to the contemporary range (Fig. 5). For the southern population, ENMs inferred suitable conditions in the southern part of the N. fuscipes range for contemporary climatic conditions, matching the current distribution of the population. The distribution of the southern population was inferred to occur even further south during the LGM (Fig. 5).

A Combined north present day; B combined north last glacial maximum; C south present day; D south last glacial maximum. Note the reduction of possible suitable areas in the past for both the northern and southern populations. Blue lines in A and C indicate rivers >50 m (made with Natural Earth. Free vector and raster map data @ naturalearthdata.com), and blue pixels in B and D indicate ice or paleolakes during the last glacial maximum (Dyke et al. 2003). The blue triangle in B and D indicate the origin of expansion for the combined north and south, respectively.
Range expansion
The range expansion analyses indicated that both population expansion origins were close to the middle of the contemporary Neotoma fuscipes range in California, near the San Francisco Bay-Delta area (Fig. 5; Supplementary Fig. 6). The north origin of expansion located at a slightly higher latitude and closer to the Sierra Nevada (p < 0.001), while the southern origin was located closer to coastal California (not significant; p > 0.1), just east of the San Francisco Bay.
Genetic diversity
Results from ThetaMater showed the southern population had the highest median posterior θ, indicating it holds the highest levels of genetic diversity (Fig. 6), followed by North A and then North B.

Note the southern population had the highest diversity, followed by North A, and then North B.
Discussion
Quaternary climate oscillations across California’s landscape had dramatic effects on patterns of biological and genetic diversity (Rissler et al. 2006). Here, we show that the dusky-footed woodrat responded to past climatic shifts through widespread contraction and expansion of its range, which led to lineage divergence and a spatial distribution of genetic variation that remains evident today. By integrating genomic data with demographic and ecological niche modeling, we were able to reconstruct the dynamic history of this species across its range.
Deep divergence and the distribution of suitable environmental conditions
Matocq (2002b) proposed that Neotoma fuscipes, and its sister species Neotoma macrotis diverged from one another ~2 million years ago in the foothills of the central Sierra Nevada, likely between Auburn and Placerville, the current distributional limits of each species (Matocq and Murphy 2007). Under this scenario, within a short window of time, early N. fuscipes would have expanded to the north and west around a partly inundated northern Central Valley and experienced a major divergence event leading to the northern and southern N. fuscipes groups (referred to as the northern and west central groups by Matocq 2002b). Our estimates of this early divergence within N. fuscipes using genome-wide SNPs dates to ~1.7 million years ago, largely consistent with the original mtDNA-based estimate of 1.8 mya (Matocq 2002b). This genetic divergence is mirrored by morphological differentiation within N. fuscipes (Hooper 1940), supporting not only the depth of this divergence but also its potential functional significance.
Fossil evidence of the history of N. fuscipes is sparse but not inconsistent with the genetic data. Neotoma fossils are widespread throughout the more southerly parts of California and the west by the Pliocene (Paleobiology DB search for “Neotoma” on 17 April 2020, paleobiodb.org), perhaps mirroring the more stable habitat available in the southern parts of the state. The earliest Neotoma fossils in the central and northern parts of the state are not found until the Irvingtonian 1.8–0.3 Ma, around the same time as or after the initial divergence events among species and between clades within N. fuscipes inferred from the genetic data. These earliest fossils are found in the San Francisco Bay Area (Alameda County; Savage 1951) and the San Joaquin Valley (Fairmead Landfill, Madera County; Dundas et al. 1996), and thus within the overall region identified as key origins of expansion for both lineages within N. fuscipes. These early fossils are not identified to individual species within Neotoma, so additional collections focused on the potential regions of divergence and subsequent expansion, aided by new methods for species-level identifications of specimens, would be extremely useful.
The central portion of California was particularly dynamic in the mid to late Pleistocene (Bartow 1991), and many factors could have contributed to the generation and maintenance of early divergence within N. fuscipes. These processes include continued and repeated glaciations extending down the western slopes of the Sierra Nevada (Richmond and Fullerton 1986), large (Sacramento River) and shifting freshwater drainages (Lock et al. 2006), extensive volcanic activity including the emergence of the Sutter Buttes in the northern Central Valley at ~1.6 mya (Prothero 2017), the inundation of the Central Valley by Corcoran Lake from ~700 to 600 ka (Bartow 1991), and the sudden establishment of the outflow of Corcoran Lake through the Carquinez Strait and what would become the modern San Francisco Bay-Delta ~600 ka (Sarna-Wojcicki et al. 1985; Bartow 1991). This historic landscape dynamism coupled with the fact that the modern San Francisco Bay-Delta presents a strong contemporary barrier to north–south movement for terrestrial species has led to this region being a concordant phylogeographic break among many taxa (Rodrı́guez-Robles JA et al. 2001; Feldman and Spicer 2006; Martínez-Solano et al. 2007; Phuong et al. 2014; Lavin et al. 2018; reviewed by Rissler et al. 2006; Gottscho 2016).
Following initial divergence of the northern and southern lineages of N. fuscipes, our demographic modeling shows there was little admixture between the two groups, with one group largely isolated to the north of the modern day San Francisco Bay–Sacramento–San Joaquin Delta, and the other largely restricted to the south, but primarily in the Coast Ranges on the western side of the Central Valley. Our niche modeling of present and LGM distributions of suitable environmental conditions for the northern and southern lineages can be viewed as rough proxies of conditions during repeated glacial (LGM) and interglacial (present) conditions of the mid to late Pleistocene (Millar and Woolfenden 2016). These models suggest that the southern lineage would have had fairly continuous access to suitable conditions in the South Coast Ranges through both glacial and interglacial times, while the northern lineage would likely have experienced much greater shifts in suitable conditions, likely leading to repeated episodes of range expansion and contraction. Our estimates of the location of lineage coalescence, or the source area for subsequent expansion, for the northern lineage is located at a central point in the Central Valley, while the source point of the southern lineage is at the northern end of the South Coast Ranges near the modern San Francisco Bay-Delta.
The northern range
The dynamic history of the northern range of N. fuscipes makes it possible that there were multiple episodes of range expansion and contraction. One or more of these episodes led to a significant divergence within the northern lineage ~76 ka where the northern Sierra Nevada meet the southern Cascade Range. This timing is potentially consistent with an early Wisconsin glaciation where there is glacial evidence (Tahoe till) in the east-central Sierra Nevada that is younger than 118–119 ka and a till from Crater Lake National Park, Oregon that is older than 67 and 72 ka (Richmond and Fullerton 1986). In addition to repeated glaciations of the northern Sierra Nevada and southern Cascades, the southern Cascades have been particularly volcanically active (Sarna-Wojcicki et al. 1985). In California, Mount Shasta and Mount Lassen along with lava flows across the Modoc Plateau have likely repeatedly influenced local to regional habitat suitability for woodrats throughout the Pleistocene and Holocene. Spatially concordant with the genetic subdivision we found at the Sierra Nevada–Cascades transition, Hooper (1940) found a morphological subdivision between the subspecies N. f. fuscipes and N. f. streatori, suggesting the potential ecological significance of this subdivision. Matocq’s (2002b) limited sampling in the northern Sierra Nevada and mtDNA analysis led to a failure to detect the distinction of the North A and North B clades.
In addition to woodrats, many other species show genetic discontinuities between the northern Sierra Nevada and southern Cascades (e.g., Rodrı́guez-Robles JA et al. 2001; Barrowclough et al. 2005; Kuchta et al. 2009; Phuong et al. 2014; Lavin et al. 2018). As in other parts of California, these common phylogeographic breaks are characterized by different depths of divergence across taxa depending on which particular glacial or volcanic event impacted a taxon in that region (Matocq 2002b). Of particular note is that spotted owls, an important predator of woodrats, have a similar distribution to N. fuscipes in northern California and share a similar spatial genetic disjunction at the boundary of the northern Sierra Nevada and southern Cascades (Barrowclough et al. 2005).
Range expansion and secondary contact
Our niche modeling suggests that the southern lineage has occupied a part of the range that potentially had suitable environmental conditions through both glacial and interglacial periods of the Quaternary. Our range expansion analysis suggests that sometime after the initial divergence with the northern lineage, the southern lineage expanded into the South Coast Range region from the modern San Francisco Bay region. The possible north to south colonization of the Inner Coast Range by this lineage of N. fuscipes is also reflected in mtDNA data, with more northern haplotypes of the Inner Coast Range ancestral to haplotypes at the southern end of the range (Matocq 2002b). Using our SNP data, our estimates of relatively large effective population size, high genetic diversity, deeper divergences among genotypes (Figs. 1, 4, and 6), and a relatively pronounced pattern of isolation by distance (Fig. 3) suggest relative stability in this portion of the range in comparison to the northern lineage. This is consistent with mtDNA data that also showed greater diversity and clade structure in the southern (west central) range in comparison to the north (Matocq 2002b). Regions inferred to be climatically stable through time are typically characterized by the accumulation of higher levels of genetic diversity and deeper genetic structure (Lessa et al. 2003; Carnaval et al. 2009; Jezkova et al. 2015, 2016; but see Hewitt 2004; Excoffier and Ray 2008), as seen here in the southern portion of the N. fuscipes range.
In contrast to the relative stability of the southern range, the northern range was likely characterized by repeated expansions and contractions of N. fuscipes, based on the known history of the region and our ENM’s for this portion of the range (Fig. 5). The relatively small effective population size (Fig. 4), low genetic diversity (Fig. 6), shallow-star-like clade structure (Fig. 1; Slatkin and Hudson 1991; Avise 2000) and weak pattern of isolation by distance (Fig. 3) suggest that modern patterns of genetic variation in northern N. fuscipes are the result of relatively recent expansion (re-expansion) into northern California. It is likely that clades North A and North B re-expanded separately into their current distributions, but our lack of thorough sampling of the North B range precludes robust modeling of this process. Additionally, these methods assume populations are in a continuous and homogenous landscape and incorporating fragmented landscapes should be explored in future studies. Nonetheless, we were able to identify an area of lineage admixture just west of the Mount Lassen area. Interestingly, here too, there is striking concordance with an important predator of N. fuscipes, the spotted owl. This is the region in which northern and California spotted owl meet, and these two lineages admix in the area near Mount Lassen, just like N. fuscipes (Barrowclough et al. 2005). These authors suggest that this area of admixture between spotted owl lineages is due to a density trough related to unsuitable habitat. In particular, much of the area is characterized by open understory pine forest, whereas owls prefer habitat with a more well-developed understory (Gutiérrez and Barrowclough 2005). Likewise, woodrats typically only occupy forested areas that have a well-developed understory in which they can build their houses (Sakai and Noon 1993; Innes et al. 2007). Interestingly, then, both predator and prey are characterized by a shared regional phylogeographic break and a concordant area of subsequent secondary contact and admixture. Space use (Carey et al. 1992; Zabel et al. 1995) and fitness (Thome et al. 1999) of spotted owls are correlated to woodrat abundance, so while both species may be responding independently to habitat structure in the area of admixture, owls are also likely responding to woodrat availability. This intriguing pattern suggests that building community-wide genomic, ENM, and demographic modeling datasets may shed light on the ecological interactions and evolutionary processes that underlie patterns of phylogeographic and population genetic concordance among taxa.
Future directions
One limitation of our work, as with most efforts to hindcast species distributions, is the use of only climatic variables in our estimations. Ideally, future modeling efforts would incorporate biotic interactions, including those with vegetation and predators, and for these woodrats in particular—their interactions with congeners. The range of N. fuscipes today and through time has been strongly influenced by the distribution of closely related congeners with which they often compete or even hybridize (Matocq 2002a, 2012; Matocq and Murphy 2007; Coyner et al. 2015; Dochtermann and Matocq 2016; Hunter et al. 2017). Woodrat species often compete for resources such as optimal nest sites and food resources (Dial 1988), leading to minimal overlap of species ranges and fine-scale parapatry (Matocq and Murphy 2007; Coyner et al. 2015; Shurtliff et al. 2014). The notion that differentially adapted woodrat species replace each other in response to changing climate conditions is evident in the paleorecord (Smith et al. 1995, 2009) and even on a multi-annual scale (Gillespie et al. 2008; Hunter et al. 2017). In northern California, we suspect the distributional response of N. fuscipes has been influenced in part by the changing distributions of its relatively cold-adapted congener, the bushy-tailed woodrat N. cinerea (Hornsby and Matocq 2012) and the relatively arid-adapted N. lepida (Gillespie et al. 2008). As stated previously, most specimens are not distinguished at the species level in the fossil record of northern California [though both N. cinerea and N. fuscipes have been identified from cave deposits in northern California (Furlong 1904; Sinclair 1907; Stock 1917)], so morphology-based work on the genus would be useful, and genetic distinction of these species and reconstruction of their changing occupancy through time could be reconstructed using ancient DNA methods (reviewed in Shapiro and Hofreiter 2014). Ecological niche modeling for N. fuscipes, like many species, will be improved with integration of ecologically and physiologically relevant parameters.
Our demographic analyses would also be improved with greater sampling, especially in the range of the North B clade and in the southern portion of the distribution. Another limitation to our study is the relatively small number of demographic scenarios we explored, given that the history and demography of N. fuscipes are certainly much more complex. For example, it is likely that these taxa, like many others, experienced asymmetric gene flow in their history. We only explored four asymmetric gene flow scenarios as a subset of scenario 11 (Supplementary Fig. 7). Our results suggest that while asymmetric models may be better (Supplementary Table 11), parameter estimates are consistent between symmetric and asymmetric models (Supplementary Table 7). Finally, the assumptions of mutation rate and generation time used in this study directly affect parameter estimates. For example, having a longer generation time or a slower mutation rate would shift the estimates of population subdivision further back in time. Nonetheless, genome-wide datasets and the potential to exploit SFS patterns is a huge advance in the field of population genetics and these data and methods will continue to be refined and integrated with even more sophisticated simulation and spatial modeling approaches that promise improved insight into complex biogeographic histories.
Data archiving
The DNA sequences used in this study are deposited at NCBI SRA (accession: PRJNA634210). All R code and data used to perform the analyses are available at https://github.com/bloispaleolab/Neotoma_demography.
References
Adams RH, Schield DR, Card DC, Corbin A, Castoe TA (2018) ThetaMater: Bayesian estimation of population size parameter θ from genomic data (Oliver, Ed.). Bioinformatics 34:1072–1073
Aiello-Lammens ME, Boria RA, Radosavljevic A, Vilela B, Anderson RP (2015) spThin: an R package for spatial thinning of species occurrence records for use in ecological niche models. Ecography 38:541–545
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Anderson RP, Raza A (2010) The effect of the extent of the study region on GIS models of species geographic distributions and estimates of niche evolution: preliminary tests with montane rodents (genus Nephelomys) in Venezuela. J Biogeogr 37:1378–1393
Avise JC (2000). Phylogeography: the history and formation of species. Harvard University Press
Badgley C, Smiley TM, Terry R, Davis EB, DeSantis LRG, Fox DL et al. (2017) Biodiversity and topographic complexity: modern and geohistorical perspectives. Trends Ecol Evol 32:211–226
Bartow JA (1991) The Cenozoic evolution of the San Joaquin Valley, California (No. 1501)
Barve N, Barve V, Jiménez-Valverde A, Lira-Noriega A, Maher SP, Peterson AT et al. (2011) The crucial role of the accessible area in ecological niche modeling and species distribution modeling. Ecol Model 222:1810–1819
Barrowclough GF, Groth JG, Mertz LA, Gutiérrez RJ (2005) Genetic structure, introgression, and a narrow hybrid zone between northern and California spotted owls (Strix occidentalis). Mol Ecol 14:1109–1120
Blois JL, McGuire JL, Hadly EA (2010) Small mammal diversity loss in response to late-Pleistocene climatic change. Nature 465:771–774
Boria RA, Olson LE, Goodman SM, Anderson RP (2014) Spatial filtering to reduce sampling bias can improve the performance of ecological niche models. Ecol Mod 275:73–77. https://doi.org/10.1016/j.ecolmodel.2013.12.012
Brown JH (1968) Adaptation to environmental temperature in two species of woodrats, Neotoma cinerea and N. albigula. Misc Publ Mus Zool Univ Mich 135:1–48
Burbrink FT, Chan YL, Myers EA, Ruane S, Smith BT, Hickerson MJ (2016) Asynchronous demographic responses to Pleistocene climate change in Eastern Nearctic vertebrates. Ecol Lett 19:1457–1467
Carey AB, Horton SP, Biswell BL (1992) Northern spotted owls: influence of prey base and landscape character. Ecol Monogr 62:223–250
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C (2009) Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science 323:785–789
Carraway LN, Verts BJ (1991) Neotoma fuscipes. Mamm Specÿies 386:1–10
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Clark PU, Shakun JD, Baker PA, Bartlein PJ, Brewer S, Brook E et al. (2012) Global climate evolution during the last deglaciation. Proc Natl Acad Sci USA 109:E1134–E1142
Coyner BS, Murphy PJ, Matocq MD (2015) Hybridization and asymmetric introgression across a narrow zone of contact between Neotoma fuscipes and N. macrotis (Rodentia: Cricetidae). Biol J Linn Soc 115:162–172
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Dansgaard W, Johnsen SJ, Clausen HB, Dahl-Jensen D, Gundestrup NS, Hammer CU et al. (1993) Evidence for general instability of past climate from a 250-kyr ice-core record. Nature 364:218–220
Davis EB, Koo MS, Conroy C, Patton JL, Moritz C (2008) The California Hotspots Project: identifying regions of rapid diversification of mammals. Mol Ecol 17:120–138
Dial KP (1988) Three sympatric species of Neotoma: dietary specialization and coexistence. Oecologia 76:531–537
Dochtermann NA, Matocq MD (2016) Speciation along a shared evolutionary trajectory. Curr Zool 62:507–511
Dundas RG, Smith RB, Verosub KL (1996) The Fairmead Landfill Locality (Pleistocene, Irvingtonian), Madera County, California: preliminary report and significance. PaleoBios 17:50–58
Dyke, A S; Moore, A; Robertson, L. Geological Survey of Canada, Open File 1574, 2003, 2 sheets; 1 CD-ROM. https://doi.org/10.4095/214399. (Open Access)
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Dupanloup I, Huerta-Sanchez E, Sousa VC, FollSousa M (2013) Robust demographic inference from genomic and SNP data. PLoS Genet 9:e1003905–e1003905
Excoffier L, Ray N (2008) Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol 23:24–28
Feldman CR, Spicer GS (2006) Comparative phylogeography of woodland reptiles in California: repeated patterns of cladogenesis and population expansion. Mol Ecol 15:2201–2222
Foley J, Rejmanek D, Foley C, Matocq M (2016) Fine-scale genetic structure of woodrat populations (Genus: Neotoma) and the spatial distribution of their tick-borne pathogens. Ticks Tick-Borne Dis 7:243–253
Furlong EL (1904) An account of the preliminary excavations in a recently explored Quaternary Cave in Shasta County, California. Science 20:53–55
Gillespie SC, Vuren DHV, Kelt DA, Eadie JM, Anderson DW (2008) Dynamics of rodent populations in semiarid habitats in Lassen County, California. West North Am Nat 68:76–82
Goldman EA (1910). Revision of the wood rats of the Genus Neotoma. U.S. Government Printing Office
Gottscho AD (2016) Zoogeography of the San Andreas Fault system: Great Pacific Fracture Zones correspond with spatially concordant phylogeographic boundaries in western North America: zoogeography of the San Andreas Fault system. Biol Rev 91:235–254
Graham RW, Lundelius EL, Graham MA, Schroeder EK, Toomey RS, Anderson E et al. (1996) Spatial response of Mammals to Late Quaternary environmental fluctuations. Science 272:1601–1606
Grivet D, Deguilloux M-F, Petit RJ, Sork VL (2006) Contrasting patterns of historical colonization in white oaks (Quercus spp.) in California and Europe. Mol Ecol 15:4085–4093
Gutiérrez RJ, Barrowclough GF (2005) Redefining the distributional boundaries of the Northern and California Spotted Owls: implications for conservation. Condor 107:182–187
Harrington SM, Hollingsworth BD, Higham TE, Reeder TW (2017) Pleistocene climatic fluctuations drive isolation and secondary contact in the red diamond rattlesnake (Crotalus ruber) in Baja California. J Biogeogr 45:64–75
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B 359:183–195
Hooper ET (1938) Geographical variation in wood rats of the species Neotoma fuscipes. University of California Press
Hooper ET (1940) Geographic variation in bushy-tailed wood rats. Univ Calif Press 42:407–424
Hopkins DM, Matthews JV, Schweger CE (2013). Paleoecology of Beringia. Elsevier
Hornsby AD, Matocq MD (2012) Differential regional response of the bushy-tailed woodrat (Neotoma cinerea) to late Quaternary climate change. J Biogeogr 39:289–305
Hotaling S, Muhlfeld CC, Giersch JJ, Ali OA, Jordan S, Miller MR et al. (2018) Demographic modelling reveals a history of divergence with gene flow for a glacially tied stonefly in a changing post-Pleistocene landscape. J Biogeogr 45:304–317
Huang H, Knowles LL (2016) Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of rad sequences. Syst Biol 65:357–365
Hunter EA, Matocq MD, Murphy PJ, Shoemaker KT (2017) Differential effects of climate on survival rates drive hybrid zone movement. Curr Biol 27:3898–3903.e4
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Hutchison DW, Templeton AR (1999) Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53:1898–1914
Innes RJ, Van Vuren DH, Kelt DA, Johnson ML, Wilson JA, Stine PA (2007) Habitat associations of dusky-footed woodrats (Neotoma fuscipes) in mixed-Conifer Forest of the Northern Sierra Nevada. J Mammal 88:1523–1531
Jackson ST, Betancourt JL, Lyford ME, Gray ST, Rylander KA (2005) A 40,000-year woodrat-midden record of vegetational and biogeographical dynamics in north-eastern Utah, USA. J Biogeogr 32:1085–1106
Jackson ST, Overpeck JT (2000) Responses of plant populations and communities to environmental changes of the late Quaternary. Paleobiology 26:194–220
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI et al. (2017) The K = 2 conundrum. Mol Ecol 26:3594–3602
Jezkova T, Jaeger JR, Oláh-Hemmings V, Jones KB, Lara-Resendiz RA, Mulcahy DG et al. (2016) Range and niche shifts in response to past climate change in the desert horned lizard Phrynosoma platyrhinos. Ecography 39:437–448
Jezkova T, Riddle BR, Card DC, Schield DR, Eckstut ME, Castoe TA (2015) Genetic consequences of postglacial range expansion in two codistributed rodents (genus Dipodomys) depend on ecology and genetic locus. Mol Ecol 24:83–97
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Jukes TH, Cantor CR (1969). Mammalian protein metabolism. Elsevier
Knowles LL, Massatti R (2017) Distributional shifts—not geographic isolation—as a probable driver of montane species divergence. Ecography 40:1475–1485
Kuchta SR, Parks DS, Mueller RL, Wake DB (2009) Closing the ring: historical biogeography of the salamander ring species Ensatina eschscholtzii. J Biogeogr 36:982–995
Lane RS, Brown RN (1991) Wood rats and Kangaroo rats: potential reservoirs of the Lyme disease Spirochete in California. J Med Entomol 28:299–302
Lapointe F, Rissler LJ (2005) Congruence, Consensus, and the Comparative Phylogeography of Codistributed Species in California. Am Nat 166:290–299
Lavin BR, Wogan GOU, McGuire JA, Feldman CR (2018) Phylogeography of the Northern Alligator Lizard (Squamata, Anguidae): hidden diversity in a western endemic. Zool Scr 47:462–476
Leaché AD, Banbury BL, Felsenstein J, De Oca ANM, Stamatakis A (2015) Short tree, long tree, right tree, wrong tree: new acquisition bias corrections for inferring SNP phylogenies. Syst Biol 64:1032–1047
Lessa EP, Cook JA, Patton JL (2003) Genetic footprints of demographic expansion in North America, but not Amazonia, during the Late Quaternary. Proc Natl Acad Sci USA 100:10331–10334
Liu Z, Otto-Bliesner BL, He F, Brady EC, Tomas R, Clark PU et al. (2009) Transient simulation of last deglaciation with a new mechanism for Bølling–Allerød Warming. Science 325:310–314
Lock J, Kelsey H, Furlong K, Woolace A (2006) Late Neogene and Quaternary landscape evolution of the northern California Coast Ranges: evidence for Mendocino triple junction tectonics. GSA Bull 118:1232–1246
Lorenz DJ, Nieto-Lugilde D, Blois JL, Fitzpatrick MC, Williams JW (2016) Downscaled and debiased climate simulations for North America from 21,000 years ago to 2100AD. Sci Data 3:160048
Martínez-Solano I, Jockusch EL, Wake DB (2007) Extreme population subdivision throughout a continuous range: phylogeography of Batrachoseps attenuatus (Caudata: Plethodontidae) in western North America. Mol Ecol 16:4335–4355
Matocq MD (2002a) Morphological and molecular analysis of a contact zone in the Neotoma fuscipes species complex. J Mammal 83:866–883
Matocq MD (2002b) Phylogeographical structure and regional history of the dusky-footed woodrat, Neotoma fuscipes. Mol Ecol 11:229–242
Matocq MD, Kelly PA, Phillips SE, Maldonado JE (2012) Reconstructing the evolutionary history of an endangered subspecies across the changing landscape of the Great Central Valley of California. Mol Ecol 21:5918–5933
Matocq MD, Murphy PJ (2007) Fine-scale phenotypic change across a species Transition Zone in the genus Neotoma: disentangling independent. Evol Phylogenet Hist Evol 61:2544–2557
Millar CI, Woolfenden WB (2016). Ecosystems past. In: Mooney H, Zavaleta E, Chapin MC (eds) Ecosystems of California. University of California Press, pp. 131–154
Muscarella R, Galante PJ, Soley-Guardia M, Boria RA, Kass JM, Uriarte M et al. (2014) ENMeval: an R package for conducting spatially independent evaluations and estimating optimal model complexity for Maxent ecological niche models. Methods Ecol Evol 5:1198–1205
Myers N, Mittermeier RA, Mittermeier CG, Fonseca GAB, da, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858
Nachman MW, Crowell SL (2000) Estimate of the mutation rate per nucleotide in humans. Genetics 156:297–304
Nogués-Bravo D, Rodríguez J, Hortal J, Batra P, Araújo MB (2008) Climate change, humans, and the extinction of the woolly mammoth. PLoS Biol 6:e79
Parisi M (ed) (2003). Atlas of the biodiversity of California. California Department of Fish and Game, Sacramento, CA
Pearson RG, Raxworthy CJ, Nakamura M, Townsend PA (2007) Predicting species distributions from small numbers of occurrence records: a test case using cryptic geckos in Madagascar. J Biogeogr 34:102–117
Peter BM, Slatkin M (2013) Detecting range expansion from genetic data. Evolution 67:3274–3289
Peter BM, Slatkin M (2015) The effective founder effect in a spatially expanding population. Evolution 69:721–734
Peterson AT (2009) Phylogeography is not enough: the need for multiple lines of evidence. Front Biogeogr 1:19–25
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 7(5):e37135. https://doi.org/10.1371/journal.pone.0037135
Phillips SJ, Anderson RP, Dudík M, Schapire RE, Blair ME (2017) Opening the black box: an open-source release of Maxent. Ecography 40:887–893
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Model 190:231–259
Phuong MA, Lim MCW, Wait DR, Rowe KC, Moritz C (2014) Delimiting species in the genus Otospermophilus (Rodentia: Sciuridae), using genetics, ecology, and morphology: Species delimitation in ground squirrels. Biol J Linn Soc 113:1136–1151
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using Multilocus Genotype data. Genetics 155:945–959
Prothero DR (2017). California’s amazing geology. CRC Press
Reid BN, Kass JM, Wollney S, Jensen EL, Russello MA, Viola EM et al. (2019) Disentangling the genetic effects of refugial isolation and range expansion in a trans-continentally distributed species. Heredity 122:441–457. https://doi.org/10.1038/s41437-018-0135-5
Reid BN, Kass JM, Wollney S, Russello ELJMA, Naro-maciel JBIMZPCJRE (2018) Disentangling the genetic effects of refugial isolation and range expansion in a trans-continentally distributed species. Heredity
Richmond GM, Fullerton DS (1986) Summation of quaternary glaciations in the United States of America. Quat Sci Rev 5:183–196
Rissler LJ, Hijmans RJ, Graham CH, Moritz C, Wake DB (2006) Phylogeographic lineages and species comparisons in conservation analyses: a case study of California Herpetofauna. Am Nat 167:655–666
Rodrı́guez-Robles JA, Stewart GR, Papenfuss TJ (2001) Mitochondrial DNA-based phylogeography of North American Rubber Boas, Charina bottae (Serpentes: Boidae). Mol Phylogenet Evol 18:227–237
Sakai HF, Noon BR (1993) Dusky-footed woodrat abundance in different-aged forests in Northwestern California. J Wildl Manag 57:373–382
Sarna-Wojcicki AM, Meyer CE, Bowman HR, Hall NT, Russell PC, Woodward MJ et al. (1985) Correlation of the Rockland Ash Bed, a 400,000-year-old stratigraphic marker in Northern California and Western Nevada, and implications for Middle Pleistocene Paleogeography of Central California. Quat Res 23:236–257
Savage D (1951) Late Cenozoic vertebrates of the San Francisco Bay region. Calif Publ Depart Geol Sci 28:1–30
Schierenbeck KA (2014). Phylogeography of California: an introduction. University of California Press
Shapiro B, Hofreiter M (2014) A paleogenomic perspective on evolution and gene function: new insights from ancient DNA. Science 343(6169):1236573. https://doi.org/10.1126/science.1236573
Shcheglovitova M, Anderson RP (2013) Estimating optimal complexity for ecological niche models: a jackknife approach for species with small sample sizes. Ecol Model 269:9–17
Shurtliff QR, Murphy PJ, Matocq MD (2014) Ecological segregation in a small mammal hybrid zone: habitat-specific mating opportunities and selection against hybrids restrict gene flow on a fine spatial scale. Evolution 68:729–742
Sinclair WJ (1907) The Exploration of the Potter Creek Cave. University of California Press
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129:555–562
Smith FA, Brown JH, Betancourt JL (1995) Evolution of body size in the woodrat over the past 25,000 years of climate change. Science 270:2012–2014
Smith FA, Crawford DL, Harding LE, Lease HM, Murray IW, Raniszewski A et al. (2009) A tale of two species: extirpation and range expansion during the late Quaternary in an extreme environment. Glob Planet Change 65:122–133
Soulé ME, Estes JA, Miller B, Honnold DL (2005) Strongly interacting species: conservation policy, management, and ethics. BioScience 55:168–176
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Stamatakis A, Ott M, Ludwig T (2005) RAxML-OMP: an efficient program for phylogenetic inference on SMPs. In: Malyshkin V (ed.) Parallel computing technologies, Lecture notes in computer science. Springer, Berlin, Heidelberg, pp. 288–302
Stock C (1917) The Pleistocene Fauna of Hawver Cave. University of California Press
Svenning J-C, Skov F (2004) Limited filling of the potential range in European tree species: limited range filling in European trees. Ecol Lett 7:565–573
Thome DM, Zabel CJ, Diller LV (1999) Forest stand characteristics and reproduction of northern spotted owls in managed North-Coastal California forests. J Wildl Manag 63:44–59
van Strien MJ, Holderegger R, Van Heck HJ (2015) Isolation-by-distance in landscapes: considerations for landscape genetics. Heredity 114:27–37
Waltari E, Hijmans RJ, Peterson AT, Nyári ÁS, Perkins SL, Guralnick RP (2007) Locating Pleistocene Refugia: comparing phylogeographic and ecological niche model predictions (J Chave, Ed.). PLoS ONE 2:e563
Whitford WG, Steinberger Y (2010) Pack rats (Neotoma spp.): Keystone ecological engineers? J Arid Environ 74:1450–1455
Zabel CJ, McKelvey K, Ward Jr. JP (1995) Influence of primary prey on home-range size and habitat-use patterns of northern spotted owls (Strix occidentalis caurina). Can J Zool 73:433–439
Acknowledgements
We thank Blois lab members, N. Byer, and C. Feldman for their helpful comments and discussion. We thank for helpful discussions and comments on a previous version of this manuscript. We also thank the editor and four anonymous reviewers for providing helpful comments. We are deeply grateful for the field collection efforts of C. Conroy, J.L. Patton, and others of the Grinnell Resurvey Project of the University of California Museum of Vertebrate Zoology, that substantially augmented samples available to this study. The authors gratefully acknowledge computing time on the Multi-Environment Computer for Exploration and Discovery (MERCED) cluster at UC Merced, funded by National Science Foundation (NSF) Grant No. ACI-1429783. RAB was supported by a Ford Predoctoral Fellowship; JLB and RAB were supported in part by the NSF (EAR-1750597); and MDM was supported in part by the NSF (IOS-1457209 and OIA-1826801).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Lounès Chikhi
Supplementary information
Rights and permissions
About this article

Cite this article
Boria, R.A., Brown, S.K., Matocq, M.D. et al. Genome-wide genetic variation coupled with demographic and ecological niche modeling of the dusky-footed woodrat (Neotoma fuscipes) reveal patterns of deep divergence and widespread Holocene expansion across northern California. Heredity 126, 521–536 (2021). https://doi.org/10.1038/s41437-020-00393-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-00393-7
