
Abstract
Comparing gene expression patterns and genetic polymorphisms between populations is of central importance for understanding the origin and maintenance of biodiversity. Based on population-specific gene expression levels and allele frequency differences, we sought to identify population divergence (PD) genes across the introgression-resistant genomic regions of Populus trichocarpa. Genes containing highly diverged loci [i.e., genetic divergence (GD)] or showing expression divergence (ED) between populations were widely distributed in the genome and substantially enriched in functional categories related to stress responses, disease resistance, timing of flowering, cell cycle regulation, plant growth, and development. Nine genomic regions showing evidence of strong positive selection were overlapped with GD genes, which had significant differences between Oregon (a southernmost peripheral deme) and the other demes. However, we did not find evidence that genes under positive selection show an enrichment for ED. PD genes and genes under selection pertained to the same gene classes, such as SERINE/CYSTEINE PROTEASE, ABC TRANSPORTER, GLYCOSYLTRANSFERASE and other transferases. Our analysis also revealed that GD genes were polymorphic within the species (41.9 ± 3.66 biallelic variants per gene), as previously reported in herbaceous plants. By contrast, ED genes contained less genetic variants (10.73 ± 1.14) and were likely highly expressed. In addition, we found that trans- rather than cis-acting variants considerably contribute to the evolution of >90% PD genes. Overall, this study elucidates that cohorts of PD genes agree with the general attributes of known speciation genes and GD genes will provide substrates for positive selection to operate on.
Similar content being viewed by others

Introduction
Understanding how genetic divergence leads to the emergence of new populations or species is of prime evolutionary interest (Seehausen et al. 2014). Gene flow and natural selection are two crucial processes accounting for how populations and/or species form (Mallet 2005; Nosil et al. 2009). By exchanging genetic material between different gene pools, gene flow influences genetic diversity including structural diversity by generating novel structural variation. Compared with the mating between individuals within species, introgression represents a long-term outcome of gene flow between species through hybridization and backcrossing. It is adaptive if foreign variants of a donor species can be maintained in the recipient species by natural selection (Anderson and Hubricht 1938). As an important evolutionary force, introgression may provide adaptive variation for the recipient species (Anderson 1953; Rieseberg and Wendel 1993) and can substantially alter the evolutionary trajectory of populations (Rieseberg et al. 1999). Introgressable genomic fragments have the potential to confer a fitness advantage by introducing foreign alleles simultaneously at multiple unlinked loci (Abbott et al. 2013; Mallet 2007). By contrast, introgression-resistant genomic blocks imply constraints in hybridizing taxa, potentially leading to reproductive isolation; one well-supported finding is reduced introgression on sex -determining regions (Barton 1979; 2001; Qvarnstrom and Bailey 2009). Moreover, elevated genomic divergence can occur in the absence of recent gene flow through divergent selection (Han et al. 2017; Renaut et al. 2014). It is, therefore, of great interest to examine how introgression-resistant genomic regions or genes in particular maintain their integrity and thus contribute to the adaptive divergence of populations and species through natural selection (Orr et al. 2004; Wu 2001; Wu and Ting 2004).
Recent studies of adaptive introgression in plants have extended to long generation taxa with “porous” species boundaries such as poplars (Chhatre et al. 2018; Stölting et al. 2013; Suarez-Gonzalez et al. 2016; 2018b). Populus hybrids are common in sympatric zones of interfertile poplar species (DiFazio et al. 2011). Evidence has shown introgression between two Populus congeners, P. trichocarpa and P. balsamifera, in northern British Columbia, Canada (Geraldes et al. 2014), leading to asymmetric adaptive introgression of disease resistance (Suarez-Gonzalez et al. 2018a; 2018b). Surprisingly, the sex chromosome in poplars appears to have experienced rampant introgression rather than the accumulation of isolation genes (Stölting et al. 2013), which is at odds with conventional knowledge on the role of sex chromosomes in speciation (Qvarnstrom and Bailey 2009). In addition to introgression, evidence has also revealed strong post-zygotic isolation (one key mechanism in speciation) due to genetic incompatibilities between Populus spp. (Lexer et al. 2010; Lindtke et al. 2014). Generally speaking, speciation is a multidimensional process involving a combination of genetic, geographic, ecological, and demographic factors (Avise 2000; Nosil et al. 2017). Understanding the continuous nature of speciation and constraints thereof (Mallet 1995) requires knowledge on the origin of alleles and/or genes under divergent or incompatibility selection (Ravinet et al. 2017; Wolf and Ellegren 2017).
Genetic introgression provides material for adaptive evolution, but it confounds our understanding of population divergence (PD). In this study, we aim to explore how divergent populations maintain their differences when challenged by introgression. We use Populus trichocarpa as a study system to examine genetic bases of PD in introgression-resistant regions of the expressed sequences. An accumulation of genetic divergence underpins the process of speciation. Along the speciation continuum, PD can be viewed as a nascent stage of speciation (Clausen 1951). In a broad-sense, a portion of population-level divergent genes likely constitute speciation genes. Here, we defined PD genes as any genes that exhibit differential gene expression (expression divergence; ED gene) or coding-region variation, namely, single nucleotide polymorphism (SNP) variants associated with allelic difference (genetic divergence; GD gene) between demes. Note that it has been demonstrated that differential expression in ED genes is able to cause adaptive phenotypic changes in animals (e.g., refs. (Abzhanov et al. 2006; Chan et al. 2010; McBride et al. 2014)); as such, they have the potential to beget PD and speciation (Haerty and Singh 2006). Nonetheless, such a causative relationship has been reported only in a few cases [e.g., ref. (Kradolfer et al. 2013) in Arabidopsis and refs. (Chung et al. 2014; Dion-Côté et al. 2014; Thomae et al. 2013) in animals]. By unifying gene expression (RNA-seq) and SNP variants (genotyping) within the coding-region sequences, this study specifically seeks to address the following questions:
-
(1)
Based on introgression-resistant loci of the transcriptome, what is the relatedness between demes at the genetic level? Does population structure inferred from these loci change compared with that obtained from genome-wide all SNP variants?
-
(2)
Is phenotypic-level population divergence driven by a few loci of major effect or many genome-wide differences (i.e., GD)? Do these divergent populations also show widespread ED in the genome?
-
(3)
How do changes in genetic variation impact changes in gene expression? Using SNP variants associated with variation in gene expression (i.e., regulatory expression quantitative trait loci [eQTLs]), are there enriched eQTLs for ED genes? Do cis- or trans-eQTLs contribute more to the evolution of PD genes?
-
(4)
Given that genes under divergent or balancing selection between populations, a priori, yield more inter-population genetic differentiation than neutral loci, do genomic regions harboring PD genes overlap with regions that have undergone positive selection? What are the biological functions for both PD genes and genes under selection? The answer to this question helps understand whether or not local adaptation (adaptive loci under positive selection) and PD have a common genetic basis.
We underpin the results of PD genes under selection by examining selective sweeps and searching for PD genes across the hitherto known introgression-resistant genomic regions in a large number of individuals from five demes across the geographical distribution range of P. trichocarpa.
Methods
Plant material and sample collection
Branch cuttings from naturally growing trees of Populus trichocarpa Torr. & A. Gray located in 29 drainages (topographic units separated by watershed barriers) extending 14° in latitude (45.6 to 59.6°N) spanning the species’ geographic distribution range (44 to 60°N, −121 to −138°W) in the Pacific Northwest were collected by British Columbia Ministry of Forests, Lands and Natural Resource Operations (Xie et al. 2009). Cuttings were rooted and outplanted at Surrey, British Columbia, Canada (49.19°N, −122.85°W, 134 masl) in 2000. In spring 2008, cuttings were collected from the Surrey site and used to establish a randomized, replicated common garden at the University of British Columbia Research Totem Field (49.26°N, −123.25°W, 82 masl) (McKown et al. 2013). Each of the 403 genotypes was replicated by 4–20 clonal ramets and these genotypes were grouped into 139 provenances within 29 drainages (Xie et al. 2009; 2012). Of these genotypes, 182 individuals were SNP-genotyped and RNA-sequenced and each individual was represented by 2–4 clonal replications. Samples of both xylem and leaves were collected at noontime over three consecutive days. Xylem samples were scraped from the glutinous layer underneath bark at ~30 cm position from the tree base and leaf samples were taken from fully uncurled leaves at the top of the canopy. Based on population structure generated by genome-wide genetic markers (Geraldes et al. 2014), we grouped these genotypes into five demes (or populations): North (wild sourced hybrids with P. balsamifera, isolated from the southern demes via physical barriers), North-South (located at the North and South separating transect), South (species core growth region with many pure P. trichocarpa individuals identified (Suarez-Gonzalez et al. 2018a), thus representing central populations), South-Oregon (in the South-to-Oregon transition), and Oregon (southernmost sampling at the species trailing edge representing marginal populations).
Variants detection and SNP genotyping
DNA was extracted from P. trichocarpa mature leaves and three different sample sets were used to call single nucleotide polymorphisms (SNPs). Genotypes were sequenced at an expected coverage ranging from 15× to 30× using the Illumina HiSeq2000 platform (Michael Smith Genome Sciences, Vancouver, BC). Short reads from the sequencing libraries were independently aligned to the P. trichocarpa genome v. 3.0 using Burrows-Wheeler Aligner v. 0.6.1-r104 with default parameters. Mate pair metadata and marked duplicate molecules were corrected using the FixMateInformation and MarkDuplicates methods in the Picard package (http://picard.sourceforge.net). Reads present in areas surrounding InDels were re-aligned using the IndelRealigner module in Genome Analysis Toolkit (GATK) v. 1.5-25-gf46f7d0. Subsequently, SNPs and small indels were independently called using the UnifiedGenotyper method from GATK. The SNPs were then filtered to exclude variants within 3 bp of any identified variants, having a mapping quality <5, and a variant quality <30. Each SNP was annotated using SNPeff (Cingolani et al. 2012) with the P. trichocarpa genome v. 3.0.
RNA-seq in stem xylem and leaves
Transcriptomic data were obtained from a population-wide RNA-sequencing using the P. trichocarpa provenances described above. Total RNA was extracted with a Purelink kit (Invitrogen, USA) and cleaned with a RNeasy Plant Mini Kit (Qiagen, Germany). Quantity and quality were checked with a 2100 BioAnalizer (Agilent, USA). Samples with RIN ≥ 9 were treated with a Nextera DNA library preparation kit (Illumina, USA) to generate 75-nt pair-end reads on the Illumina HiSeq2000 platform. Short reads were trimmed off adaptor sequences and checked for quality using Trimmomatic v.0.32 (Bolger et al. 2014) (minimum quality 20 over 4-bp sliding window, minimum length of 50 bp) before alignment to the P. trichocarpa genome v. 3.0 using TopHat v.2.0.8 (Trapnell et al. 2012) (mean inner distance 300 bp with 20-bp standard deviation between reads). Reads mapped to multiple loci of the genome were given a mapping quality of zero. The RNA-seq data, presented as fragments per kilobase of transcript per Million mapped reads (FPKM), were calculated using Cufflinks v. 2.1.1 (Trapnell et al. 2012). A total of 26,741 and 27,986 primary annotated gene transcripts was yielded in the xylem and leaf RNA-seq data, respectively. After excluding genes over swathes of introgressed genome regions [information from ref. (Suarez-Gonzalez et al. 2018a)], we used 26,328 and 27,548 genes in the xylem and leaf transcriptomic data, respectively, for all subsequent analyses.
Population stratification, genetic estimates, and individual genetic relationship
Population structure was estimated using PLINK v. 1.90 (Purcell et al. 2007) based on 172,408 SNPs distributed in the introgression-resistant transcriptomic regions across 19 chromosomes. The maximum cluster size parameter was set to 5 (--mc 5). Linkage disequilibrium (LD) for each deme and individual inbreeding coefficients (F) were calculated also using PLINK v. 1.90 through the “--r2” and “--het” command option, respectively (Purcell et al. 2007). To characterize population differentiation between demes and genetic diversity across the introgression-resistant transcriptomic regions of each deme, population genetic parameters (Nei’s nucleotide diversity (π), Tajima’s D, and Hudson’s FST) were calculated in a 1-bp sliding window with a step size of 1 bp on PopGenome v. 2.2.4 (Pfeifer et al. 2014). Further, we constructed a species phylogenetic tree using a coalescent model, implemented by SNAPP v. 1.4.2 (Bryant et al. 2012), a package of the Bayesian software BEAST v. 2.5.2 (Bouckaert et al. 2019). Owing to the computational demands of analyzing the entire dataset, we reduced the number of individuals in the South deme by choosing all pure individuals and at least one individual from each drainage with priority given to those with the least missing data. After this selection step, every deme had a balanced number of individuals (ca. 14 individuals per deme) and a total of 65 individuals was retained. Based on biallelic SNPs within genes (N = 122,672), we only used those with no missing data (N = 15,411) for analysis. The starting mutation rate parameters were calculated directly from the sequence alignment (u = 1.213, v = 0.851). The default value 10 was used for the “coalescent rate” parameter and the value of the parameter was sampled (estimated in the Markov chain Monte Carlo [MCMC]). The priors for ancestral population sizes were chosen to be a relatively broad gamma distribution with parameters α = 2 and β = 200. A Yule tree prior with λ = 0.01 and a gamma prior for θ (α = 11.75, β = 109.73) were used. Three separate MCMC runs of 106 generations were performed, sampling every 103 generations. We assessed convergence using Tracer v. 1.7.1 (Rambaut et al. 2018) and used LogCombiner to combine the runs after removing 10% of the trees as a burn-in. The MCMC traces for each run were shown in Supplementary Fig. S7. The BEAST tree topology was visualized as a cloudogram with Densitree v. 2.2.5 (Bouckaert 2010).
Detection of genetic loci of genomic population divergence
We identified highly differentiated genomic loci for each deme using a Hidden Markov Model (HMM) approach (Hofer et al. 2012; Marques et al. 2016; Soria-Carrasco et al. 2014). In brief, the HMM is based on three hidden states, that is, genomic background (assumedly neutral under a hierarchical island model), regions of exceptionally low divergence, and regions of exceptionally high divergence. We used highly diverged regions as loci of PD. The most likely state of each SNP was inferred from the HMM, based on observed Hudson’s FST values for each deme. We optimized parameters of the HMM from 1000 random initial parameters using the Baum-Welch algorithm (Baum et al. 1970) implemented in the R package HiddenMarkov v.1.8.11. The best HMM parameter was used to reconstruct the most likely states using the Viterbi algorithm (Viterbi 1967). R script to perform the HMM was available at https://github.com/marqueda/HMM-detection-of-genomic-islands.
Expression level population divergence based on Q ST and fold-change
To test population differentiation in gene expression, we estimated levels of gene expression divergence based on QST—the quantitative genetic equivalent of FST (Leinonen et al. 2013; McKay and Latta 2002). QST was estimated following the method of (Spitze 1993). The additive variance components between (\(\sigma _{{\mathrm{between}}}^2\)) and within (\(\sigma _{{\mathrm{within}}}^2\)) populations for each gene were obtained through nested analysis of variance (i.e., drainages nested within demes) using the MCMC approach in the R package MCMCglmm v.2.29 (Hadfield 2010). In a Bayesian framework, each model employed resampling strategies across individuals within population. Specifically, we used inverse Wishart priors and an MCMC of 50,000 iterations with a burn-in of 10%. QST was calculated as \(\sigma _{{\mathrm{between}}}^2/\left( {\sigma _{{\mathrm{between}}}^2 + {\mathrm{2}}\sigma _{{\mathrm{within}}}^2} \right)\). To overcome the overestimation of QST, we only considered the genes with significant heritability. We calculated repeatability as an assumed upper bound estimate of broad-sense heritability (H2) of gene expression from the variance estimates based on \(\sigma _{\mathrm{G}}^2/\left( {\sigma _{\mathrm{G}}^2 + \sigma _{\mathrm{E}}^2} \right)\), where \(\sigma _{\mathrm{G}}^2\)is the genetic component of the variance estimated as the expression variance between genotypes for a given gene (i.e., variance among individual means) and \(\sigma _{\mathrm{E}}^2\) is the environmental component of the variance estimated as the expression variance within genotypes for a given gene (i.e., the mean variance among replicates). Point estimates of H2 were obtained using the repeatability function in the R package heritability v.1.2 (Kruijer et al. 2015). The significance of H2 was estimated by using an empirical null model where heritabilities were based on random genotype assignments. For each gene, 1000 permutations were performed and empirical P-values were calculated using empPvals function in the R package q-value v.2.18.0 (Bass et al. 2015).
Further, differential gene expression between demes was calculated using a quasi-likelihood negative binomial generalized log-linear model (glmQLFit), implemented in the R package edgeR v. 3.28.1 (Robinson et al. 2010). In each case of paired comparisons, genes with a false-discovery rate (FDR) corrected for multiple testing using the method of Benjamini and Hochberg below 0.05 and a fold‐change >1.2 were considered significantly differentially expressed.
Transcriptome-wide scan for selective sweeps
Genome-wide scans can identify differentiated loci between populations/species that may have promoted population/species divergence. Most of the available approaches for selection analysis are more sensitive to signatures of strong positive selection, namely, hard-selective sweeps. We used likelihood-, LD-, and FST-based approaches to detect SNPs under selection across the regions of the introgression-resistant transcriptome. We first scanned for target loci of recent positive selection using SweepFinder2 v. 1.0 (DeGiorgio et al. 2016; Nielsen et al. 2005). This software program implements the test for sweeps based on local deviations of the folded site frequency spectrum. The spacing parameter -g was set to 10 kb and the program was run for each of the five demes. The composite likelihood ratio (CLR) was estimated for each SNP and the top 100 peak-sweep signals with a CLR higher than the introgression-resistant transcriptome-wide ca. 97% quantile were used to define sweep candidates. Consequently, significance cutoffs of 26.71, 10.65, 21.63, 16.93, and 15.03 were used for North, North-South, South, South-Oregon, and Oregon demes, respectively. Second, we computed the ω statistic (LD-based) for selective sweeps at 10-kb intervals using OmegaPlus v. 2.3.0 (Alachiotis et al. 2012). The minwin and maxwin parameters were set to 100 and 1000, respectively. Similarly, the top 100 highest ω statistic values were used to select sweep candidates, which equaled to use the statistic thresholds of 10.65, 6.84, 13.79, 87, and 17.58 for the aforementioned five demes, respectively. The last approach we used was based on the expected distribution of FST (Chi-square distribution if no selection or neutral loci) implemented via OutFLANK v. 0.2 (Whitlock and Lotterhos 2015). We used the entire SNP data and generated the FST distributions after removal of the loci within the top and bottom 5% of the distribution, with an expected heterozygosity <0.01, or an FDR (q-value) >0.05. The overlap regions of the three methods showing significant selection were deemed confident-selective sweep regions. Genes within (or adjacent to) these regions were deemed genes under selection.
Multiple factor analysis
We used a multidimensional exploratory approach—multiple factor analysis (MFA)—to jointly analyze the structure emerging from one genomic and two transcriptomic data (stem xylem and leaves). A priori, the MFA is a PCA applied to each group of variables in merged data tables (i.e., each data table composed of a group of different variables but the same set of individuals across data tables). Then each variable is weighted by the first eigenvalue of the matrix of variance-covariance associated with the group it belongs to (details in Supplementary Note S1; refs. (de Tayrac et al. 2009; Pagès 2002)). The rationale of the scaling is to balance the influence from each of the groups of variables and thus the first component of each group has the same inertia. After the global analysis for the whole data, MFA provides a balanced representation of each individual by using the whole data table and a partial representation of each individual by using each of the groups of variables. The corresponding graphical displays (individual factor map and variables representation) can be read as in PCA. MFA also permits looking for specific and common structures from each variables group through a representation of each matrix of variables (groups representation). By integrating biological information as supplementary groups of variables, we are able to identify the biological processes that best reflect the molecular underpinnings (e.g., allelic frequency of genetic variants, differential gene expression) characterizing the differences between clusters for individuals. It is worth noting that these supplementary groups of variables do not participate in the construction of the dimensions (axes) of the MFA. The interpretation of the results is made through the projection of biological processes onto the dimensions from MFA (Supplementary Note S1). We used the \({\cal{L}}\)g measure (akin to the multiple correlation coefficient R2 with limits between 0 and 1) to represent and compare the groups. We performed an MFA using the R package FactoMineR v. 2.3 (Lȇ et al. 2008).
Finally, to test for whether PD genes had different gene expression or allele frequencies between two main deme clusters identified by Dims of the MFA, we calculated maximal information coefficients (MIC) using the R package minerva v. 1.5.8 (Albanese et al. 2013). This test is similar to permutation test to establish statistical significance by using resampling strategies, but has the merit of identifying both linear and nonlinear dependencies between variables. For each allele frequency- or expression-deme test, a null distribution of MIC values was created by using 500,000 bootstrapped MIC tests with randomly associated data points. P-values were calculated by determining the proportion of null MIC values that were higher than the actual counterparts; small P-values indicate less likely to get the true MIC value than expected by chance. We corrected P-values using the Benjamini–Hochberg procedure (Benjamini and Hochberg 1995) and calculated an FDR using the R package q-values v. 2.18.0 (Bass et al. 2015). Both adjusted P-values and q-values < 0.05 were considered statistically significant.
eQTL mapping
To test whether the expression of ED genes has associated SNPs, eQTL association mapping was performed using the R package MatrixEQTL v. 2.3 (Shabalin 2012). The population structure (i.e., all individuals stratified into five demes) was used as a covariate in the linear regression model implemented by MatrixEQTL. We performed the testing of the model based on t-statistic and significance threshold was set at P < 1e-6 and q-values < 0.05. cis-eQTLs typically arise due to variation in cis-regulatory elements, whereas trans-eQTLs mostly occur because of polymorphism on trans-regulatory factors located far away from the target genes (Rockman and Kruglyak 2006). We determined an eQTL as cis (e.g., promoters, enhancers and changes in homolog expression) if gene-SNP pairs are within the distance of 1Mbp; all eQTLs that did not meet this criterion were defined as trans (e.g., transcriptional factors and chromatin modifiers). Note that in this study, we considered only SNP variants in coding and regulatory regions (i.e., genes ± 800 bp), so all the cis-/ trans-eQTLs identified were within genes and their near up-/down-streams.
Functional annotation and GO enrichment
Gene ontology (GO) enrichment for ED genes was calculated using the R package topGO v. 2.36.0 (Alexa and Rahnenführer 2010). Mapping Populus trichocarpa gene identifiers to GO terms was accomplished by searching the GO database (http://geneontology.org/), which was used to customize GO annotations for subsequent use. These GOs were visualized using REVIGO (Supek et al. 2011). Of the 400 ED genes (i.e., top 100 most contributable to each of Dim 1, 2, 4, and 6 of the MFA), 46 had GO terms, and using top 10% ED genes based on contribution ratios in the MFA (see Supplementary Fig. S16), 6 were used to predefine interesting genes (parameter “geneSectionFun”). The parameter nodeSize was set to 5 to prune GO terms with fewer than five annotated genes. The weight algorithm with Fisher’s exact test was used to rank most important GOs in the enrichment analysis.
Results
Introgression-resistant transcriptome-wide genetic diversity and population-level genetic divergence
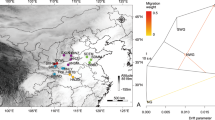
Based on genome-wide markers (Supplementary Fig. S1), the population stratification indicated that the individuals can be divided into five demes, that is, North, North-South, South, South-Oregon, and Oregon (inset of Fig. 1a). Using SNP markers within the transcribed sequences along the introgression-resistant genomic regions (track “g” of Fig. 2a), multidimensional scale (MDS) plot showed that Oregon was a genetically distinct deme with a contrasting population structure (Fig. 1a). Inbreeding coefficient (F) in Oregon and North-South was significantly different to each of the other demes (all P < 0.05 based on Wilcoxon test; Supplementary Fig. S6a) and these two demes had the highest and lowest average F values, respectively (Supplementary Fig. S6a). Overall LD decay was ca. 10–15 kb in all but South-Oregon deme, where Oregon decayed most quickly (Fig. 1b). Such strikingly fast and slow LD decays in Oregon and South-Oregon, respectively, across chromosomes (Fig. 1b) were concordant with that in most of each chromosome (Supplementary Fig. S2). We additionally note that estimate of r2 between SNP markers considerably increased when the pairwise distance increased to 250–500 kb. This increase was mainly due to 106 highly correlated SNP marker pairs (r2 > 0.45), which were found to be within 18 genes distributed in seven chromosomes (track “h” of Fig. 2a and Supplementary Table S1), suggesting that the nine gene pairs are highly correlated through genetic linkage, despite no clear functional connection (Supplementary Table S1).

Color coding for each deme is the same throughout this study. a Multidimensional scaling (MDS) plot based on pairwise identity-by-state distance for the introgression-resistant transcriptome. An inset graph shows drainages colored by deme based on Supplementary Fig. S1. b Linkage disequilibrium (LD) difference between demes. LD decays quickly within 10–15 kb for all demes but South-Oregon. All the four demes but South-Oregon have similar LD decay patterns. A horizontal dashed line marks r2 dropping to 0.2. c, d nucleotide diversity (π) and Tajima’s D for each deme (shown by chromosome in Supplementary Figs. S3 and S4, respectively). Average values displayed in boxes of the two plots. *** for significance at P < 0.0001 by Wilcoxon test. e BEAST phylogenetic tree for the transcriptome along the introgression-resistant regions. f Percentage of differentially expressed (DE) genes between demes were based on fold-changes > 2 and FDR < 5% using edgeR. Top-left and bottom-right triangle matrix for leaves and xylem data, respectively.

In graph (a), tracks from outside to inside are: a Chromosome ideogram for the P. trichocarpa genes (construction detailed in Supplementary Note S2). b and c Gene expression in stem xylem and leaves, respectively. d–f Results of selective sweeps averaged over the demes using three approaches, namely, SweepFinder2, OmegaPlus, and OutFLANK, respectively. Significant loci shown in red. g Introgressed genomic regions from a previous study (see the Methods section). h Link ribbons showing the genes (Supplementary Table S2) with r2 > 0.45 and pairwise distances between 250 and 500 kb. Graph (b) delineates the distribution of SNPs within genes ± 800 bp (N = 172,408 SNPs) across the transcribed introgression-resistant regions.
Nucleotide diversity (π) was significantly different between demes (all P < 0.05 by Wilcoxon test; Fig. 1c and Supplementary Fig. S3) and lowest in Oregon (0.0026) relative to the other demes (0.0028 on average) (Fig. 1c and Supplementary Fig. S3). Tajima’s D was significantly different in the paired comparisons between demes (all P < 0.05 by Wilcoxon test; Fig. 1d and Supplementary Fig. S4) and the South deme had the highest Tajima’s D (0.708 vs. 0.281 for an average over the other demes; Fig. 1d and Supplementary Fig. S4). By contrast, the lowest and highest Hudson’s FST values were found in South and Oregon, respectively (all P < 0.05 by Wilcoxon text; Supplementary Figs. S5 and S6b).
Furthermore, we applied a phylogenomic method, the Bayesian SNPP, to unveil individual relationships (Fig. 1e). The result showed that each of the five demes was an independent “clade” and there was a close relationship between Oregon and South-Oregon individuals (Fig. 1e).
Population-level expression divergence
Results of transcriptomic analysis showed that stem xylem gene expression was overall higher than leaf counterparts (tracks “b–c” of Fig. 2a). There were 81.5% and 95% genes expressed in xylem and leaves, respectively, that had significantly higher heritability (H2) than expected by chance (P < 0.05 based on permutation tests) (Supplementary Fig. S8a). This indicates that the expression of a large proportion of genes is under considerable genetic control. Point estimates of QST (s.d.) were 0.069 (0.27) and 0.009 (0.15) for xylem and leaf gene expression, respectively (Supplementary Fig. S8b). Based on xylem data, QST had relatively strong positive correlations with median expression (Pearson’s r = 0.32, df = 11,630, P < 2.2e-16) and expression variance (r = 0.11, df = 11,630, P < 2.2e-16). Similarly, QST based on leaf data was positively correlated with median expression (r = 0.124, df = 21,140, P < 2.2e-16) and expression variance (r = 0.066, df = 21,140, P < 2.2e-16). This indicates that genes showing expression level population divergence are more likely to be highly expressed and such a relationship is more pronounced in xylem than in leaves. For the significant genes in xylem, H2 ranged from 0.12 to 1.0 with a mean (s.d.) of 0.42 (0.2) and 8006 genes (31%) had H2 > 0.5. There was a weak negative correlation between H2 and median expression-level (Pearson’s r = −0.076, df = 26,135, P < 2.2e-16) and expression variance (Pearson’s r = −0.018, df = 26,135, P = 0.003). In leaves, H2 ranged from 0.12 to 1.0 with a mean (s.d.) of 0.53 (0.17) and 18,654 genes (58%) had H2 > 0.5. There was a weak non-significant positive correlation of H2 with median expression (Pearson’s r = 0.003, df = 31,956, P = 0.578) and a negative correlation with expression variance (Pearson’s r = −0.006, df = 31,956, P = 0.258). This indicates that highly expressed genes in xylem are under less strong genetic control than genes expressed at low or intermediate levels.
With respect to differentially expressed (DE) genes between demes, xylem had more DE genes than leaves (lower triangle vs. upper triangle matrix of Fig. 1f), concordant with the QST results (Supplementary Fig. S8b). North and North-South had few DE genes in both xylem and leaves (<2.5%; Fig. 1f and Supplementary Fig. S9); however, these two demes had high DE genes relative to the other demes in xylem (averagely ca. 25.5%; Fig. 1f and Supplementary Fig. S9). In leaves, South-Oregon had the highest DE genes relative to North-South (15.3%; Fig. 1f and Supplementary Fig. S9); South-Oregon was different from all the other demes in gene expression of both xylem and leaves (Supplementary Fig. S6c, d).
Level of population divergence and selective sweeps
We investigated how introgression-resistant genes/loci have been impacted by natural selection in each deme. We first characterized levels and heterogeneity of deme divergence across these regions using Hudson’s FST for each deme, based on a Hidden Markov model (HMM) approach. Of 172,131 SNPs used, 19.69%, 36.01%, 14.85%, 24.19%, and 23.16% had significantly high genetic divergence at α = 0.05 for North, North-South, South, South-Oregon, and Oregon, respectively. This result particularly highlighted the substantial difference between North-South and the other demes. Distribution of genetic differentiation and specific loci were showcased in Supplementary Fig. S10 and on the top of each stacked panel in Supplementary Fig. S11, respectively. Moreover, these genetic loci were overlapped with candidate regions under positive selection based on three approaches, that is, SweepFinder2, OmegaPlus, and OutFLANK (Supplementary Fig. S11). Consistently, regions with low density of genetic divergence SNPs had few positive selection loci detected (Supplementary Fig. S11). While composite likelihood ratios (CLR) from SweepFinder2 were significantly different in South-Oregon relative to the other demes (P < 0.05 by Duncan’s test; Supplementary Table S2a), ω statistics from OmegaPlus showed that each deme was significantly different to the other demes (all P < 0.05 by Duncan’s test; Supplementary Table S2b).
Selective scan identified 50 loci under strong positive selection (Supplementary Figs. S11, S12 and tracks “d–f” of Fig. 2a), whereby 55 genes were deemed under positive selection (Table 1). Those loci under strong selection were mainly detected in North and North-South (see the “Identified Deme” column of Table 1). The biological functions of these genes under selection chiefly included SERINE/CYSTEINE PROTEASE (Potri.004G147800, Potri.006G021600, Potri.006G057400, Potri.008G147600, Potri.014G120600, and Potri.019G124700), SERINE/THREONINE-PROTEIN KINASE (STK; Potri.004G147800), ABC TRANSPORTER (Potri.006G126100, Potri.008G012500, and Potri.015G063400), GLYCOSYLTRANSFERASE (Potri.006G131000, Potri.011G025700, and Potri.012G094600) and other transferases (Potri.002G186800, Potri.003G159200, Potri.003G216900, and Potri.015G101100), hydrolases (Potri.002G081300, Potri.007G095200, Potri.007G145800, and Potri.010G253700), epigenetics-related genes such as chromatin modification (Potri.004G080900) and methylation (Potri.005G179700 and Potri.007G146000), and genes related to RNA and DNA synthesis (Potri.001G055000, Potri.003G216800, Potri.006G021500, Potri.010G066700, and Potri.015G041800) (Table 1).
Identification of population divergence genes
To probe genes and/or genetic markers that are able to distinguish demes, termed PD genes or genetic markers, we applied a multivariate factor analysis (MFA) to the introgression-resistant transcriptome and SNPs within the introgression-resistant transcribed regions. In total, we used 172,408 SNPs, 26,741 and 27,986 genes (RNA-seq) expressed in stem xylem and leaves, respectively, from 151 individuals. Given the SNP-containing genes (i.e., candidate genes for GD), each gene had 31.8 SNPs on average (Supplementary Fig. S13 and Fig. 2b).
We focused on the first six principal components of the MFA (13.6% of the total variance; Supplementary Fig. S14a) for the mean representation of the individuals based on xylem and leaf transcriptomes and SNPs (Supplementary Fig. S15). Partial representations associated with each deme were obtained from the consensus between transcriptomes and SNPs points of view (i.e., transcriptome and allelic frequency variation; Fig. 3a). Each deme was represented by four points: the consensus among the three points of view (SNPs, xylem and leaf transcriptomes) and a point for each of the three points of view. Each principal component (or dimension; Dim) partitioned the individuals into grouped demes (circled in Supplementary Fig. S15c). Specifically, North and North-South were well separated from the other demes along the Dim 1; the other Dims partitioned Oregon and/or South-Oregon from the other demes (Fig. 3a and Supplementary Fig. S15). Xylem gene expression and SNPs contributed dominantly to the first six principal components (Dim 1–6; Supplementary Fig. S14b). This was concordant with partial representation and groups representation (Fig. 3b). Both representation plots consistently showed that the partition existed (i) on Dim 1 and 2 at the xylem gene expression-level and (ii) at the SNPs level on Dim 3-6 (Fig. 3). Although Dim 4 and 6 were specific to SNPs (Fig. 3), the top 100 elements most contributable to respective principal components were xylem and leaf gene expression, respectively (Supplementary Figs. S16 and S17).

In a, the balanced representation of each deme is the barycenter of the points summarizing partial points of view linked through colored lines. The projection of partial representations for leaf gene expression and SNPs of the five demes onto Dim 1 are very close, which means that leaf gene expression and SNPs define similar structures for demes on Dim 1; but this projection for xylem gene expression of the five demes is not the case: North (N) and North-South (N_S) are negative (coordinate close to −2) while the other demes (S, S_O, and O for South, South-Oregon, and Oregon, respectively) are positive (close to 1). Dim 1 is, therefore, specific to the xylem gene expression point of view that differentiates North and North-South from the other demes. On the Dim 2, the mean South-Oregon individuals from the partial xylem gene expression representation is positive (around 2 in the coordinate) while such a representation for the other demes is negative (0 to −0.5). Dim 2 is also specific to the xylem gene expression allowing to distinguish South-Oregon from the other demes. This is confirmed by analyzing the groups representation in b: only xylem gene expression has a coordinate value close to 1 (max. value) on Dim 1 and 2. Likewise, Dim 3–6 are specific to SNPs, which provide a partition of Oregon (and South-Oregon) with the other demes.
We defined the top 100 elements, either DE genes or genetic markers with different allele frequency, along the first six principal components as ED genes or GD markers based on the partition of deme groups in the MFA (shown in Supplementary Fig. S16). We found that 70.5% of ED genes had no allelic polymorphism (Supplementary Table S3). Moreover, the density of SNP variants within ED genes (10.37 ± 1.14 counts per gene) was significantly lower than that in GD marker-containing genes, namely, GD genes (41.9 ± 3.66 counts per gene) (P = 6.47e-13 by t-test; Supplementary Table S3). This indicates that there may be few cis-eQTLs within many ED genes per se and that ED genes may be regulated by trans-eQTLs. Indeed, based on our eQTL discovery, we found that trans-eQTLs were significantly more abundant than cis-eQTLs in ED genes (averagely 50 vs. 0.3 counts per gene and P < 2.2e-16 by t-test; Fig. 4a). In particular, trans-eQTLs for ED genes from Dim 1 were highly enriched than for those from the other Dims (182 vs. 6 ± 3 counts per gene; Supplementary Table S3), whereas the number of cis-eQTLs was similarly low in all ED genes from different Dims (0.26 ± 0.23 counts per gene; Supplementary Table S3). The pattern of more trans- than cis-eQTLs in ED genes was also found in all genes from both xylem and leaves (37 vs. 6 counts per gene and P < 2.2e-16 by t-test; Fig. 4a and Supplementary Fig. S18). Abundant trans-eQTLs for ED suggest that cross-talks among genes in their signaling network may be crucial to gene expression and regulation. Expression patterns of ED genes classified via a hierarchical clustering (Fig. 4b) were in line with the partition of the demes (Fig. 3a and Supplementary Fig. S15c), in which Dim 4 accounting for the separation of South-Oregon and Oregon from the demes contained many highly expressed genes (average log2FPKM ≈ 6.39 vs. 4.14 ± 0.98 for the other three Dims; Fig. 4b). Maximal information coefficients (MIC) tests showed that all the 100 ED genes from Dim 1 were statistically significant (adjusted P < 0.05 and q-value < 0.05; Fig. 4c and Supplementary Table S3), but not all for ED genes from Dim 2, 4, and 6 (Fig. 4c and Supplementary Table S3). This agreed with the clustering, which showed that Dim 1 had the lowest height (dissimilarity) of the dendrogram between broken-line demarcated deme(s) vs. the others (Fig. 4b). Moreover, we found the ED genes were not enriched in specific gene ontology (GO) terms (all P ≥ 0.05; Supplementary Table S4), and the two leading GO biological processes were involved in the regulation of transcription and cellular metabolic process (Fig. 4d). Nonetheless, there were functional differences for ED genes from Dim 1 vs. Dim 2, 4, and 6 (Supplementary Fig. S21). Compared to the former (Supplementary Fig. S21a), the latter (Supplementary Fig. S21b) had more GO biological processes such as aromatic and cyclic compound metabolisms.

a cis- and trans-eQTLs identified in all genes (N = 26,328 and 27,548 for xylem and leaves, respectively) and ED genes. b Heatmap of the expression of ED genes based on Dim 1, 2, 4, and 6 of the MFA. Ward’s method for maximum distance used for clustering. c Adjusted P-value based on the MIC test against fold-change in gene expression for two-group stratified demes on each Dim shown in a. Red dots are ED genes of each Dim and horizontal dashed lines represent significance at α = 0.05. d Top five GO terms identified by the weight algorithm for down-weighting genes in the GO enrichment. Boxes indicate the five most important terms, where filled color represents the relative significance with dark red and light yellow for most and least significant, respectively. No color ellipses for the rest of relatively less important GO terms. Black arrows indicate is-a relationships. Note that all P-values > 0.05 suggest no significant difference among the GO terms for candidate ED genes.
Clustering the GD markers, which partitioned Oregon from the other demes, consistently showcased that all the individuals of Oregon were clustered in one group (Supplementary Fig. S19). These markers were within loci of PD and some of them were in regions under strong positive selection (Fig. 5a). Moreover, all GD loci under selection were statistically significant at α = 0.05 (Fig. 5b). However, clustering the expression of GD genes from Dim 3 and 5 (Supplementary Fig. S20) demonstrated no clear separation of Oregon from the other demes as shown in the MFA (Fig. 3a).

a Loci identified by Dim 3 and 5 of the MFA distinguish Oregon from the other demes. Red asterisks indicate loci under strong positive selection only for Oregon and/or South-Oregon (more detail in Table 1). Regions with very low π and high FST in Oregon relative to the other demes (Supplementary Figs. S3 and S5) shaded in light gray in chr01, 02, and 15. Loci showing PD across demes or in Oregon based on the HMM are marked at the top (in black) or bottom (in gray) of each panel, respectively. PD genes were also marked in black dots. b P-values were calculated using the MIC test for allele frequencies between Oregon and the other demes. The horizontal red line marks the significance at α = 0.05.
Biological functions of population divergence genes
Overall, there were some common biological functions of ED genes, such as abiotic and biotic stress responses (e.g., NAC-domain-containing protein, protease, WRKY), flowering timing control (e.g., FLC), regulators of cell cycle, plant growth and development (Table 2). Of the 400 candidate ED genes, 19 encoded SERINE/THREONINE-PROTEIN KINASE, 15 were RIBOSOMAL PROTEINS, and 12 were transporters (e.g., ABC TRANSPORTER) (Supplementary Table S3). We also identified genes involved in the biosynthesis of essential amino acid, such as histidine (Potri.008G137900 and Potri.012G102800), intracellular protein-protein interactions such as F-BOX PROTEIN (Potri.002G248300, Potri.003G112000, Potri.004G157000, Potri.006G212600, and Potri.017G090300) and ankyrin repeat-containing protein (Potri.009G070700), epigenetics such as chromatin modification (Potri.002G243400 and Potri.010G174900), methylation (Potri.001G264800, Potri.001G342300, Potri.005G170800, and Potri.006G120200), and sugar-transferring enzyme such as GLYCOSYLTRANSFERASE (Potri.006G238900) (Supplementary Table S3). Moreover, we found that only ca. 12% of the ED genes were transcription factors, such as bHLH (Potri.007G112800, Potri.010G130000, Potri.013G001300, Potri.013G025900, and Potri.015G105200), MYB (Potri.011G122900), and WRKY (Potri.011G087900) (Supplementary Table S3), in line with the assumption that the genetic bases of many evolved changes are mutations within the cis-acting regulatory elements of genes (Britten and Davidson 1971; Stern 2000). These transcription factors were found to have roles in the regulation of gene expression in response to abiotic stress (Schluttenhofer and Yuan 2015; Yamaguchi-Shinozaki and Shinozaki 2005). In addition, many ED genes separating North and North-South from the other demes (i.e., Dim 1) had functions in stress responses and disease resistance; by contrast, regulators of plant growth and cell cycle abounded in ED genes differentiating Oregon and/or South-Oregon from the other demes (i.e., Dim 2, 4, and 6) (Supplementary Table S3).
Through GD markers from Dim 3 and 5, we identified their corresponding genes, namely, GD genes. The function of these genes chiefly comprised CYSTEINE SYNTHASE (Potri.013G127800), ASPARTATE or SERINE PROTEASE (Potri.006G232400 and Potri.019G131100), HEAT SHOCK TRANSCRIPTION FACTOR in stress resistance (Potri.006G226800 and Potri.015G141100), sugar-transferring enzyme such as GLYCOSYLTRANSFERASE (Potri.001G136800, Potri.005G255000, and Potri.016G042100), RIBOSOMAL PROTEIN (Potri.001G415400 and Potri.016G019000), INTRACELLULAR PROTEIN-PROTEIN INTERACTIONS (Potri.014G149400), and TRANSPORTER (Potri.010G194300 and Potri.011G150600) (Supplementary Table S3). Notably, many transporters have been found to be related to stress responses such as Na+/K+ or Na+/Ca2+ transporter in salt tolerance (Ren et al. 2005; Wang et al. 2012).
By overlapping PD genes with the genes under selection, we found that nine GD genes but no ED gene were under positive selection (genes highlighted in bold in Table 1 and in red in Supplementary Table S3). These overlapped genes were mainly identified in Dim 3 of the MFA, which can distinguish Oregon from the other demes (Figs. 3 and Supplementary Fig. S14c). The biological functions of these overlapped genes were involved primarily in proteases, disease resistance (e.g., STK of nucleotide binding sites and leucine-rich repeats (NBS-LRR) proteins), transferases and transporters (possibly related with stress response), and mRNA splicing (Table 1).
Characteristics of divergence genes between pure and hybrid individuals
Based on a whole-genome local ancestry analysis (Suarez-Gonzalez et al. 2018b), individuals from North and North-South were highly introgressed by P. balsamifera (Fig. 6a). According to this analysis for the 151 individuals used in this study, 22 pure P. trichocarpa individuals were identified and they were originated from either North or South demes (Fig. 6b). By comparing pure and hybrid individuals (N = 22 vs. 129) based on the transcriptomic and SNP data using MIC tests, we identified statistically differentially expressed genes and genetic markers (Supplementary Fig. S22), similar to ED and GD genes identified between demes, respectively. However, different to between-deme ED genes, these divergence genes were overall expressed at a low level (FPKM = 0.68 ± 0.42) but had similar functions as between-deme PD genes (Table 2), such as disease resistance (Potri.017G104800), SERINE/THREONINE-PROTEIN KINASE (Potri.007G128400 and Potri.019G120300), sugar-transferase (GLYCOSYLTRANSFERASE: Potri.007G140700 and Potri.016G052500), transporter (Potri.014G078000), phytohormone signal transduction (Potri.004G117700 and Potri.010G165700), and methylation (Potri.015G039000) (Fig. 6c). Moreover, one “GD” gene with biological function in transferase was overlapped with an identified between-deme GD gene (Potri.003G216900; Fig. 6d and highlighted in yellow in Supplementary Table S3).

a The ancestry assignment is based on a whole-genome local ancestry analysis for these individuals. b According to the ancestry analysis, 22 pure P. trichocarpa individuals (N = 151) were identified, in which 10 and 12 individuals were originated from North and South demes, respectively. c, d Using the MIC test, we probed 10 genes with significant differential expression (adjusted P < 0.05 and absolute fold-change > 2) and six genes each with ≥10 SNPs having significant difference in allele frequency (P < 0.05). See Supplementary Fig. S22 for details. Gene functions are briefly described beneath each panel. Allele frequency ⊂ {0, 1, 2}.
Discussion
Understanding the emergence and maintenance of new populations or species has long fascinated ecologists and evolutionary biologists and is a major endeavor for the evolutionary biology community. In this study, we identified PD genes that contribute to the differentiation between demes of Populus trichocarpa. As accumulation of these genes, phenotypes may change and the process of evolution would drive PD and have the potential to beget new populations and/or species. Our findings highlight that ED genes allowed to separate North and North-South from the other demes likely due to their physical barriers (Xie et al. 2009) and that both GD and ED genes enabled to distinguish Oregon and/or South-Oregon from the other demes likely due to reproductive barriers caused by their significantly different flowering timing (Liu, Ingvarsson and El-Kassaby, unpubl. Work). PD genes are widespread in the genome and have functions in stress responses and disease resistance, flowering timing, cell cycle regulation, plant growth and development (Table 2). Nine GD genes were found to be under strong positive selection (Table 1). While GD genes exhibited high intraspecific polymorphisms (Supplementary Table S3), consistent with findings from herbaceous plant species (Bomblies and Weigel 2007; Christie and Macnair 1987; Sweigart et al. 2007), ED genes harbored less polymorphic sites within the species and were likely highly expressed. In contrast to evidence accrued to show that cis-regulatory changes contribute to the response to positive selection and to ED [e.g., (de Meaux 2018; Wray 2007) but see (Bell et al. 2013)], we did not discover ED genes under positive selection and found that most of these PD genes (>90%) were trans-eQTLs rather than cis-eQTLs (Supplementary Table S3). Finally, it is worth noting that our consideration only in genes ±800 bp regions excluded the possibility of probing eQTLs in introns and intergenic sequences, although this does not undermine the finding of much more trans-eQTLs than cis-QTLs.
Oregon, a southernmost deme, likely under the strongest selection
Patterns in genomic diversity could be caused by recent adaptation through selective sweeps and divergent selection could act to favor population-specific alleles. A selective sweep caused by divergent selection between habitats is expected to entrain excess differentiation (e.g., high FST) between populations at and around the site under selection, as well as reduced diversity in the population experiencing selective sweep and strongly negative Tajima’s D upon completion of the sweep. Moreover, genomic regions under strong positive selection are expected to have lower diversity and high LD. Here, we reported that South had the lowest portion of loci showing population differentiation based on the HMM. Consistently, we found that South had the lowest Hudson’s FST (Supplementary Fig. S6b), the highest nucleotide diversity π (Fig. 1c), very positive Tajima’s D (Figs. 1d and Supplementary Fig. S4), and moderate LD decay (Fig. 1b). The geographical region where the South deme is located corresponds the place where this species preferably grows (DeBell 1990), indicating that South has likely been exposed to the environment with the least selective pressures. By contrast, Oregon likely represents the deme that has the highest Hudson’s FST (Supplementary Figs. S6b and S5), the lowest nucleotide diversity (Fig. 1c), more loci with negative Tajima’s D (Fig. 1d), and the quickest LD decay, making Oregon considerably different from the other demes in population structure and individual clustering (Fig. 1a, b, e). Altogether, we infer that South and Oregon may have undergone the weakest and strongest divergent selection, respectively. Nonetheless, we cannot rule out the possibility that population demographic histories (e.g., bottlenecks) could result in such patterns of π, Tajima’s D, and LD decay. Indeed, we have found that North and South demes have undergone more recent bottlenecks than Oregon (Liu, Ingvarsson and El-Kassaby, unpubl. Work). We therefore cannot assertively claim that the Oregon deme has undergone the most intensive selection.
Insights into population divergence genes
Using both transcriptomic and SNP data, the MFA yielded a shortlist of candidate PD genes (Supplementary Table S3). Given the assumption that some PD genes have the potential to emerge as speciation genes, reproductive isolation as a crucial mechanism, leading to speciation is related possibly with PD. Reproductive isolation is typically categorized according to the timing of isolation (pre- vs. post-zygotic isolation). Prezygotic isolation prevents the formation of hybrids via hybrid incompatibility or sexual isolation, while post-zygotic isolation renders hybrids sterile or inviable due to reduced viability or fitness of hybrids such as outbreeding depression (Orr and Presgraves 2000; Ridley 2004). Noteworthy candidate ED genes had biological functions associated with reproductive isolation. For example, many genes were involved in stress responses and disease resistance, flowering timing, regulation of cell cycle (e.g., cell division), plant growth and development (Table 2). Hybrid necrosis is a well-known reproduction isolation mechanism, caused by the spontaneous activation of plant defenses associated with leaf necrosis, stunted growth and reduced fertility in hybrids (Bomblies et al. 2007). Some causal genes of hybrid necrosis have been identified in previous studies and confirmed in this study (Supplementary Table S3), such as disease resistance genes (e.g., LEUCINE-RICH REPEAT PROTEINS (LRR)) (Bomblies et al. 2007; Dixon et al. 1996; Kruger et al. 2002) and their interacting factors [e.g., STRUBBELIG RECEPTOR FAMILY (SRF) kinase (Alcázar et al. 2010)], and cell cycle-related genes (Mizuno et al. 2011; Świadek et al. 2017). Moreover, changes in a CASEIN KINASE (serine/threonine-selective enzyme) sequence in Oryza sativa cultivars led to hybrid necrosis (Yamamoto et al. 2010). Reciprocal silencing of duplicated histidine synthesis genes (HISTIDINE KINASE) in Arabidopsis caused arrested embryo development, resulting in seed abortion (Bikard et al. 2009). Mutations of a flowering repressor, FLOWERING LOCUS C, caused delayed flowering in Arabidopsis allopolyploids (Wang et al. 2006), leading to reproductive isolating barriers (Lowry et al. 2008). Our data also suggest that epigenetic mechanisms are a possible cause for hybrid incompatibilities such as changes in DNA methylation via METHYLTRANSFERASE (Supplementary Table S3). This is accordant with a finding of lethality in Arabidopsis due to hypermethylation in a histidine biosynthesis gene (Blevins et al. 2017). We additionally singled out GLYCOSYLTRANSFERASE (a sugar-transferring enzyme) as a candidate PD gene (Supplementary Table S3), which had been found to be a target of rapid evolution and interspecific differentiation in Populus spp. (Caseys et al. 2015).
Based on candidate GD markers, we found their corresponding genes, namely, GD genes (Dim 3 and 5 in Supplementary Table S3). Interestingly, a portion of GD genes had the same function as ED genes, such as HEAT SHOCK TRANSCRIPTION FACTOR in stress response, SERINE PROTEASE in disease resistance, and GLYCOSYLTRANSFERASE (Supplementary Table S3). Moreover, there were GD markers in CYTOCHROME P450 MONOOXYGENASE, a gene that is important for the biosynthesis of defense molecules in plants (Durst and Benveniste 1993). In addition, there were other genes also found to be associated with reproductive isolation, including CYSTEINE/ASPARTATE PROTEASE, RIBOSOMAL PROTEIN, and INTRACELLULAR PROTEIN-PROTEIN INTERACTIONS (Table S3). Specifically, mutated cysteine protease in tomato led to hybrid necrosis (Kruger et al. 2002; Rooney et al. 2005) and changes in an aspartate protease protein sequence in Oryza sativa cultivars led to hybrid female sterility (Chen et al. 2008). Reciprocal silencing of duplicated nuclear-encoded mitochondrial ribosomal proteins in rice led to hybrid male sterility (Yamagata et al. 2010). Amino acid substitution of a protein-protein interaction protein in Oryza sativa resulted in hybrid male sterility (Long et al. 2008).
In addition, we found that lots of ED genes contained few variant sites (i.e., no SNPs; Supplementary Table S3), which points to the character of gene expression evolution when populations (or species) diverge and split. It has been suggested that gene expression evolution is under a combination of stabilizing selection and neutral evolution (Gilad 2012; Romero et al. 2012), which correspond to expression patterns showing small or large expression variance within and between species (or populations), respectively (Whitehead and Crawford 2006). Our results indicate that neutral evolution likely affects patterns of many DE genes between demes of P. trichocarpa. Moreover, many trans-eQTLs discovered in ED genes (Supplementary Table S3) suggest the important role of trans-regulation effects in ED. Nonetheless, trans-eQTLs are likely to be selected against due to great deleterious pleiotropic effects (Metzger et al. 2016; Wittkopp 2005; Wittkopp et al. 2004).
Genes under positive selection overlapped with genetic divergence genes
To date, few cases have documented whether there are shared genetic bases of local adaptation and PD or speciation; only one notable example in Mimulus demonstrated genetically linked loci for local adaptation to copper mine soils and hybrid lethality (Wright et al. 2013). A single adaptive allele sweeps through population, namely, hard sweep, potentially resulting in differential gene expression and/or allele frequency between demes. By aligning the PD genes and loci under positive selection along the introgression-resistant transcribed regions, we found that 50 genetic markers were under strong positive selection, which were located in 55 genes (Table 1 and Supplementary Fig. S3). These genes have functions in, for example, disease resistance (e.g., proteases, STK) (Table 1 and Supplementary Fig. S3). In addition to positive selection by fixing beneficial alleles, balancing selection also helps maintain advantageous genetic variation within genes involved in disease resistance or host-pathogen interactions in populations or species of both plants (Karasov et al. 2014; Roux et al. 2013; Wang et al. 2020) and animals (Klein et al. 2007; Leffler et al. 2013). Several recent studies further indicate that genes under balancing selection are important for local adaptation (Ma et al. 2018; Wang et al. 2020), also as evidenced by the relatedness between the distribution of divergent alleles and niche diversification (Wu et al. 2017). Our finding additionally suggests that genes under positive selection play an important role in PD and thus local adaptation as well.
Conclusion
Gene flow is able to foster the merger of different gene pools between interfertile species. Analyses of PD genes—genes that contribute to adaptive divergence of populations (e.g., reproductive isolation)—can offer clues regarding the ecological settings, evolutionary/selective forces, molecular mechanisms that drive the divergence of populations and/or species. By excluding introgressable transcriptomic regions, we identified candidate ED and GD genes in introgression-resistant regions between demes of Populus trichocarpa. Biological functions of PD genes point to their involvement in stress responses, disease resistance, timing of flowering, cell cycle regulation, plant growth and development. We found that no ED but GD genes showed evidence of significant positive selection, suggesting that GD genes evolve as a product of local adaptation to the environment. Moreover, GD genes were polymorphic, contrasting with less genetic variants in ED genes. ED genes are overall more likely to be highly expressed, especially in xylem compared to in leaves. In addition, we found that trans-regulatory changes rather than cis-acting variants substantially contribute to the evolution of PD genes. Collectively, this work demonstrates the molecular underpinnings of PD and provide evolutionary insights into adaptive divergence through the evolution of genes and expression regulations thereof.
Data availability
The original genotyping data have been deposited on SRA under the accession PRJNA276056. The RNA-sequencing data have been deposited on SRA under the accession PRJNA300564. All the intermediate data supporting the findings of this study are available upon reasonable request.
Code availability
Custom Python scripts have been made available as a notebook appendix in the Supporting Information.
References
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N et al. (2013) Hybridization and speciation. J Evol Biol 26(2):229–246
Abzhanov A, Kuo WP, Hartmann C, Grant BR, Grant PR, Tabin CJ (2006) The calmodulin pathway and evolution of elongated beak morphology in Darwin’s finches. Nature 442(7102):563–567
Alachiotis N, Stamatakis A, Pavlidis P (2012) OmegaPlus: A scalable tool for rapid detection of selective sweeps in whole-genome datasets. Bioinformatics 28(17):2274–2275
Albanese D, Filosi M, Visintainer R, Riccadonna S, Jurman G, Furlanello C (2013) minerva and minepy: A C engine for the MINE suite and its R, Python and MATLAB wrappers. Bioinformatics 29(3):407–408
Alcázar R, García AV, Kronholm I, de Meaux J, Koornneef M, Parker JE et al. (2010) Natural variation at strubbelig receptor kinase 3 drives immune-triggered incompatibilities between Arabidopsis thaliana accessions. Nat Genet 42:1135
Alexa A, Rahnenführer J (2010) topGO: enrichment analysis for gene ontology. R package version 2.28.0. https://bioconductor.org/packages/topGO/
Anderson E (1953) Introgressive hybridization. Biol Rev Camb Philos Soc 28(3):280–307
Anderson E, Hubricht L (1938) Hybridization in Tradescantia. III The evidence for introgressive hybridization. Am J Bot 25(6):396–402
Avise J (2000) Phylogeography: The history and formation of species. Harvard University Press, Cambridge, MA
Barton N (1979) The dynamics of hybrid zones. Heredity 43:341–359
Barton NH (2001) The role of hybridization in evolution. Mol Ecol 10(3):551–568
Bass AJ, Dabney A, Robinson D (2015). qvalue: Q-value estimation for false discovery rate control. R package version 2.10.0. http://github.com/jdstorey/qvalue.
Baum LE, Petrie T, Soules G, Weiss N (1970) A maximization technique occurring in statistical analysis of probabilistic functions of markov chains. Ann Math Stat 41(1):164–171
Bell GDM, Kane NC, Rieseberg LH, Adams KL (2013) RNA-seq analysis of allele-specific expression, hybrid effects, and regulatory divergence in hybrids compared with their parents from natural populations. Genome Biol Evol 5(7):1309–1323
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B Met 57(1):289–300
Bikard D, Patel D, Le Mette C, Giorgi V, Camilleri C, Bennett MJ et al. (2009) Divergent evolution of duplicate genes leads to genetic incompatibilities within A. thaliana. Science 323(5914):623–626
Blevins T, Wang J, Pflieger D, Pontvianne F, Pikaard CS (2017) Hybrid incompatibility caused by an epiallele. Proc Natl Acad Sci USA 114(14):3702–3707
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Bomblies K, Lempe J, Epple P, Warthmann N, Lanz C, Dangl JL et al. (2007) Autoimmune response as a mechanism for a Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol 5:1962–1972
Bomblies K, Weigel D (2007) Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nat Rev Genet 8:382
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A et al. (2019) BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comp Biol 15(4):e1006650
Bouckaert RR (2010) DensiTree: Making sense of sets of phylogenetic trees. Bioinformatics 26(10):1372–1373
Britten RJ, Davidson EH (1971) Repetitive and non-repetitive DNA sequences and a speculation on the origins of evolutionary novelty. Q Rev Biol 46(2):111–138
Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, RoyChoudhury A (2012) Inferring species trees directly from biallelic genetic markers: Bypassing gene trees in a full coalescent analysis. Mol Biol Evol 29(8):1917–1932
Caseys C, Stritt C, Glauser G, Blanchard T, Lexer C (2015) Effects of hybridization and evolutionary constraints on secondary metabolites: The genetic architecture of phenylpropanoids in European populus species. PLoS ONE 10(5):e0128200
Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, Brady SD et al. (2010) Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327(5963):302–305
Chen JJ, Ding JH, Ouyang YD, Du HY, Yang JY, Cheng K et al. (2008) A triallelic system of S5 is a major regulator of the reproductive barrier and compatibility of indica-japonica hybrids in rice. Proc Natl Acad Sci USA 105(32):11436–11441
Chhatre VE, Evans LM, DiFazio SP, Keller SR (2018) Adaptive introgression and maintenance of a trispecies hybrid complex in range-edge populations of Populus. Mol Ecol 27(23):4820–4838
Christie P, Macnair MR (1987) The distribution of postmating reproductive isolating genes in populations of the yellow monkey flower, Mimulus guttatus. Evolution 41(3):571–578
Chung H, Loehlin DW, Dufour HD, Vaccarro K, Millar JG, Carroll SB (2014) A single gene affects both ecological divergence and mate choice in. Drosoph Sci 343(6175):1148–1151
Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6(2):80–92
Clausen J (1951) Stages in the evolution of plant species Cornell University Press, Ithaca, New York
de Meaux J (2018) Cis-regulatory variation in plant genomes and the impact of natural selection. Am J Bot 105(11):1788–1791
de Tayrac M, Lê S, Aubry M, Mosser J, Husson F (2009) Simultaneous analysis of distinct Omics data sets with integration of biological knowledge: multiple Factor Analysis approach. BMC Genomics 10(1):32
DeBell DS (1990) Populus trichocarpa Torr. & Gray, black cottonwood, vol 2. US Department of Agriculture, Forest Service, Agriculture Handbook, Washington, D.C., USA
DeGiorgio M, Huber CD, Hubisz MJ, Hellmann I, Nielsen R (2016) SweepFinder2: Increased sensitivity, robustness and flexibility. Bioinformatics 32(12):1895–1897
DiFazio SP, Slavov GT, Joshi CP (2011) Populus: A premier pioneer system for plant genomics. In: Joshi C, Difazio SP, Kole C (eds). Genetics, genomics and breeding of poplar. Science Publishers, Enfield, NH
Dion-Côté A-M, Renaut S, Normandeau E, Bernatchez L (2014) RNA-seq reveals transcriptomic shock involving transposable elements reactivation in hybrids of young lake whitefish species. Mol Biol Evol 31(5):1188–1199
Dixon MS, Jones DA, Keddie JS, Thomas CM, Harrison K, Jones JDG (1996) The tomato Cf-2 disease resistance locus comprises two functional genes encoding leucine-rich repeat proteins. Cell 84(3):451–459
Durst F, Benveniste I (1993) Cytochrome P450 in plants, vol 105. Springer, Berlin, Heidelberg
Geraldes A, Farzaneh N, Grassa CJ, McKown AD, Guy RD, Mansfield SD et al. (2014) Landscape genomics of Populus trichocarpa: the role of hybridization, limited gene flow, and natural selection in shaping patterns of population structure. Evolution 68(11):3260–3280
Gilad Y (2012) Using genomic tools to study regulatory evolution. Humana Press, New York
Hadfield JD (2010) MCMC methods for multi-response generalized linear mixed models: The MCMCglmm R package. J Stat Softw 1:2
Haerty W, Singh RS (2006) Gene regulation divergence is a major contributor to the evolution of Dobzhansky-Muller incompatibilities between species of Drosophila. Mol Biol Evol 23(9):1707–1714
Han F, Lamichhaney S, Grant BR, Grant PR, Andersson L, Webster MT (2017) Gene flow, ancient polymorphism, and ecological adaptation shape the genomic landscape of divergence among Darwin’s finches. Genome Res 27(6):1004–1015
Hofer T, Foll M, Excoffier L (2012) Evolutionary forces shaping genomic islands of population differentiation in humans. BMC Genomics 13(1):107
Karasov TL, Kniskern JM, Gao LP, DeYoung BJ, Ding J, Dubiella U et al. (2014) The long-term maintenance of a resistance polymorphism through diffuse interactions. Nature 512(7515):436–440
Klein J, Sato A, Nikolaidis N (2007) MHC, TSP, and the origin of species: From immunogenetics to evolutionary genetics. Annu Rev Genet 41:281–304
Kradolfer D, Wolff P, Jiang H, Siretskiy A, Kohler C (2013) An imprinted gene underlies postzygotic reproductive isolation in Arabidopsis thaliana. Dev Cell 26(5):525–535
Kruger J, Thomas CM, Golstein C, Dixon MS, Smoker M, Tang SK et al. (2002) A tomato cysteine protease required for Cf-2-dependent disease resistance and suppression of autonecrosis. Science 296(5568):744–747
Kruijer W, Boer MP, Malosetti M, Flood PJ, Engel B, Kooke R et al. (2015) Marker-based estimation of heritability in immortal populations. Genetics 199(2):379–398
Lȇ S, Josse J, Husson F (2008) FactoMineR: an R package for multivariate analysis. J Stat Softw 25(1):1–18
Leffler EM, Gao Z, Pfeifer S, Ségurel L, Auton A, Venn O et al. (2013) Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 339(6127):1578–1582
Leinonen T, McCairns RJ, O’Hara RB, Merilä J (2013) QST-FST comparisons: evolutionary and ecological insights from genomic heterogeneity. Nat Rev Genet 14(3):179–190
Lexer C, Joseph JA, van Loo M, Barbará T, Heinze B, Bartha D et al. (2010) Genomic admixture analysis in European Populus spp. reveals unexpected patterns of reproductive isolation and mating. Genetics 186(2):699–712
Lindtke D, Gompert Z, Lexer C, Buerkle CA (2014) Unexpected ancestry of Populus seedlings from a hybrid zone implies a large role for postzygotic selection in the maintenance of species. Mol Ecol 23(17):4316–4330
Long YM, Zhao LF, Niu BX, Su J, Wu H, Chen YL et al. (2008) Hybrid male sterility in rice controlled by interaction between divergent alleles of two adjacent genes. Proc Natl Acad Sci USA 105(48):18871–18876
Lowry DB, Modliszewski JL, Wright KM, Wu CA, Willis JH (2008) The strength and genetic basis of reproductive isolating barriers in flowering plants. Philos T R Soc B 363(1506):3009–3021
Ma T, Wang K, Hu Q, Xi Z, Wan D, Wang Q et al. (2018) Ancient polymorphisms and divergence hitchhiking contribute to genomic islands of divergence within a poplar species complex. Proc Natl Acad Sci USA 115(2):E236–43
Mallet J (1995) A species definition for the modern synthesis. Trends Ecol Evol 10(7):294–299
Mallet J (2005) Hybridization as an invasion of the genome. Trends Ecol Evol 20(5):229–237
Mallet J (2007) Hybrid speciation. Nature 446(7133):279–283
Marques DA, Lucek K, Meier JI, Mwaiko S, Wagner CE, Excoffier L et al. (2016) Genomics of rapid incipient speciation in sympatric threespine stickleback. PLoS Genet 12(2):e1005887
McBride CS, Baier F, Omondi AB, Spitzer SA, Lutomiah J, Sang R et al. (2014) Evolution of mosquito preference for humans linked to an odorant receptor. Nature 515(7526):222–227
McKay JK, Latta RG (2002) Adaptive population divergence: Markers, QTL and traits. Trends Ecol Evol 17(6):285–291
McKown AD, Guy RD, Azam MS, Drewes EC, Quamme LK (2013) Seasonality and phenology alter functional leaf traits. Oecologia 172(3):653–665
Metzger BPH, Duveau F, Yuan DC, Tryban S, Yang B, Wittkopp PJ (2016) Contrasting frequencies and effects of cis- and trans-regulatory mutations affecting gene expression. Mol Biol Evol 33(5):1131–1146
Mizuno N, Shitsukawa N, Hosogi N, Park P, Takumi S (2011) Autoimmune response and repression of mitotic cell division occur in inter-specific crosses between tetraploid wheat and Aegilops tauschii Coss. that show low temperature-induced hybrid necrosis. Plant J 68(1):114–128
Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C (2005) Genomic scans for selective sweeps using SNP data. Genome Res 15(11):1566–1575
Nosil P, Feder JL, Flaxman SM, Gompert Z (2017) Tipping points in the dynamics of speciation. Nat Ecol Evol 1(2):0001
Nosil P, Funk DJ, Ortiz-Barrientos D (2009) Divergent selection and heterogeneous genomic divergence. Mol Ecol 18(3):375–402
Orr HA, Masly JP, Presgraves DC (2004) Speciation genes. Curr Opin Genet Dev 14(6):675–679
Orr HA, Presgraves DC (2000) Speciation by postzygotic isolation: Forces, genes and molecules. Bioessays 22(12):1085–1094
Pagès J (2002) Analyse factorielle multiple appliquée aux variables qualitatives et aux données mixtes. Rev Statistique Appliquée 4:5–37
Pfeifer B, Wittelsbürger U, Ramos-Onsins SE, Lercher MJ (2014) PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol Biol Evol 31(7):1929–1936
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2007) PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575
Qvarnstrom A, Bailey RI (2009) Speciation through evolution of sex-linked genes. Heredity 102(1):4–15
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67(5):901–904
Ravinet M, Faria R, Butlin RK, Galindo J, Bierne N, Rafajlović M et al. (2017) Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. J Evol Biol 30(8):1450–1477
Ren Z-H, Gao J-P, Li L-G, Cai X-L, Huang W, Chao D-Y et al. (2005) A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat Genet 37(10):1141–1146
Renaut S, Owens GL, Rieseberg LH (2014) Shared selective pressure and local genomic landscape lead to repeatable patterns of genomic divergence in sunflowers. Mol Ecol 23(2):311–324
Ridley M (2004) Evolution. Blackwell Publishing, Oxford, UK
Rieseberg L, Wendel J (1993) Introgression and its consequences in plants. Oxford University Press, New York, NY
Rieseberg LH, Archer MA, Wayne RK (1999) Transgressive segregation, adaptation and speciation. Heredity 83:363–372
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140
Rockman MV, Kruglyak L (2006) Genetics of global gene expression. Nat Rev Genet 7(11):862–872
Romero IG, Ruvinsky I, Gilad Y (2012) Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet 13(7):505–516
Rooney HCE, van’t Klooster JW, van der Hoorn RAL, Joosten MHAJ, Jones JDG, de Wit PJGM (2005) Cladosporium Avr2 inhibits tomato Rcr3 protease required for Cf-2-dependent disease resistance. Science 308(5729):1783–1786
Roux C, Pauwels M, Ruggiero MV, Charlesworth D, Castric V, Vekemans X (2013) Recent and ancient signature of balancing selection around the S-locus in Arabidopsis halleri and A. lyrata. Mol Biol Evol 30(2):435–447
Schluttenhofer C, Yuan L (2015) Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol 167(2):295–306
Seehausen O, Butlin RK, Keller I, Wagner CE, Boughman JW, Hohenlohe PA et al. (2014) Genomics and the origin of species. Nat Rev Genet 15:176
Shabalin AA (2012) Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 28(10):1353–1358
Soria-Carrasco V, Gompert Z, Comeault AA, Farkas TE, Parchman TL, Johnston JS et al. (2014) Stick insect genomes reveal natural selection’s role in parallel speciation. Science 344(6185):738–742
Spitze K (1993) Population structure in Daphnia obtusa: quantitative genetic and allozymic variation. Genetics 135(2):367–374
Stern DL (2000) Evolutionary developmental biology and the problem of variation. Evolution 54(4):1079–1091
Stölting KN, Nipper R, Lindtke D, Caseys C, Waeber S, Castiglione S et al. (2013) Genomic scan for single nucleotide polymorphisms reveals patterns of divergence and gene flow between ecologically divergent species. Mol Ecol 22(3):842–855
Suarez-Gonzalez A, Hefer CA, Christe C, Corea O, Lexer C, Cronk QC et al. (2016) Genomic and functional approaches reveal a case of adaptive introgression from Populus balsamifera (balsam poplar) in P. trichocarpa (black cottonwood). Mol Ecol 25(11):2427–2442
Suarez-Gonzalez A, Hefer CA, Lexer C, Cronk QCB, Douglas CJ (2018a) Scale and direction of adaptive introgression between black cottonwood (Populus trichocarpa) and balsam poplar (P.balsamifera). Mol Ecol 27(7):1667–1680
Suarez-Gonzalez A, Hefer CA, Lexer C, Douglas CJ, Cronk QCB (2018b) Introgression from Populus balsamifera underlies adaptively significant variation and range boundaries in P. trichocarpa. N Phytol 217(1):416–427
Supek F, Bošnjak M, Škunca N, Šmuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6(7):e21800
Sweigart AL, Mason AR, Willis JH (2007) Natural variation for a hybrid incompatibility between two species of Mimulus. Evolution 61(1):141–151
Świadek M, Proost S, Sieh D, Yu J, Todesco M, Jorzig C et al. (2017) Novel allelic variants in ACD6 cause hybrid necrosis in local collection of Arabidopsis thaliana. N Phytol 213(2):900–915
Thomae AW, Schade GOM, Padeken J, Borath M, Vetter I, Kremmer E et al. (2013) A pair of centromeric proteins mediates reproductive isolation in Drosophila species. Dev Cell 27(4):412–424
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7(3):562–578
Viterbi AJ (1967) Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. IEEE Trans Inf Theory 13(2):260–269
Wang JL, Tian L, Lee HS, Chen ZJ (2006) Nonadditive regulation of FRI and FLC loci mediates flowering-time variation in Arabidopsis allopolyploids. Genetics 173(2):965–974
Wang M, Zhang L, Zhang Z, Li M, Wang D, Zhang X et al. (2020) Phylogenomics of the genus Populus reveals extensive interspecific gene flow and balancing selection. N Phytol 225(3):1370–1382
Wang P, Li Z, Wei J, Zhao Z, Sun D, Cui S (2012) A Na+/Ca2+ exchanger-like protein (AtNCL) involved in salt stress in Arabidopsis. J Biol Chem 287(53):44062–44070
Whitehead A, Crawford DL (2006) Neutral and adaptive variation in gene expression. Proc Natl Acad Sci USA 103(14):5425–5430
Whitlock MC, Lotterhos KE (2015) Reliable detection of loci responsible for local adaptation: inference of a null model through trimming the distribution of FST. Am Nat 186:S24–S36
Wittkopp PJ (2005) Genomic sources of regulatory variation in cis and in trans. Cell Mol Life Sci 62(16):1779–1783
Wittkopp PJ, Haerum BK, Clark AG (2004) Evolutionary changes in cis and trans gene regulation. Nature 430(6995):85–88
Wolf JBW, Ellegren H (2017) Making sense of genomic islands of differentiation in light of speciation. Nat Rev Genet 18(2):87–100
Wray GA (2007) The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8(3):206–216
Wright KM, Lloyd D, Lowry DB, Macnair MR, Willis JH (2013). Indirect evolution of hybrid lethality due to linkage with selected locus in Mimulus guttatus. PLoS Biol 11(2):e1001497
Wu CI (2001) The genic view of the process of speciation. J Evol Biol 14(6):851–865
Wu CI, Ting CT (2004) Genes and speciation. Nat Rev Genet 5(2):114–122
Wu Q, Han TS, Chen X, Chen JF, Zou YP, Li ZW et al. (2017) Long-term balancing selection contributes to adaptation in Arabidopsis and its relatives. Genome Biol 18(1):217
Xie C-Y, Ying CC, Yanchuk AD, Holowachuk DL (2009) Ecotypic mode of regional differentiation caused by restricted gene migration: A case in black cottonwood (Populus trichocarpa) along the Pacific Northwest coast. Can J For Res 39(3):519–525
Xie CY, Carlson MR, Ying CC (2012) Ecotypic mode of regional differentiation of black cottonwood (Populus trichocarpa) due to restricted gene migration: Further evidence from a field test on the northern coast of British Columbia. Can J For Res 42(2):400–405
Yamagata Y, Yamamoto E, Aya K, Win KT, Doi K, Sobrizal et al. (2010) Mitochondrial gene in the nuclear genome induces reproductive barrier in rice. Proc Natl Acad Sci USA 107(4):1494–1499
Yamaguchi-Shinozaki K, Shinozaki K (2005) Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci 10(2):88–94
Yamamoto E, Takashi T, Morinaka Y, Lin SY, Wu JZ, Matsumoto T et al. (2010) Gain of deleterious function causes an autoimmune response and Bateson-Dobzhansky-Muller incompatibility in rice. Mol Genet Genomics 283(4):305–315
Acknowledgements
This work was supported by funds of the Genome Canada Large-Scale Applied Research Program (POPCAN; project 168BIO) to YAE (and other leading PIs of the project team: C. J. Douglas, R. D. Guy, S. D. Mansfield, and Q. C. B. Cronk), and by a National Sciences and Engineering Research Council of Canada Discovery Grant to YAE. We thank Q. C. B. Cronk (UBC) for assistance in RNA-seq data pre-processing. We are equally grateful for Compute Canada (Cedar system) to afford part of computational simulations. Finally, we thank the Editor and anonymous referees for their helpful comments after thorough reading and granting an extended time for revision due to the COVID pandemic.
Author information
Authors and Affiliations
Contributions
YAE led and coordinated the project (experimental design and data collection); YL conceived of this study, performed data analyses, and wrote the manuscript with supportive inputs from YAE. All authors read and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Liu, Y., El-Kassaby, Y.A. Transcriptome-wide analysis of introgression-resistant regions reveals genetic divergence genes under positive selection in Populus trichocarpa. Heredity 126, 442–462 (2021). https://doi.org/10.1038/s41437-020-00388-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-00388-4
