
Abstract
Understanding the ecological and evolutionary processes occurring during species range shifts is important in the current context of global change. Here, we investigate the interplay between recent expansion, gene flow and genetic drift, and their consequences for genetic diversity and structure at landscape and local scales in European beech (Fagus sylvatica L.) On Mont Ventoux, South-Eastern France, we located beech forest refugia at the time of the most recent population minimum, ~150 years ago, and sampled 71 populations (2042 trees) in both refugia and expanding populations over an area of 15,000 ha. We inferred patterns of gene flow and genetic structure using 12 microsatellite markers. We identified six plots as originating from planting, rather than natural establishment, mostly from local genetic material. Comparing genetic diversity and structure in refugia versus recent populations did not support the existence of founder effects: heterozygosity (He = 0.667) and allelic richness (Ar = 4.298) were similar, and FST was low (0.031 overall). Still, significant spatial evidence of colonization was detected, with He increasing along the expansion front, while genetic differentiation from the entire pool (βWT) decreased. Isolation by distance was found in refugia but not in recently expanding populations. Our study indicates that beech capacities for colonization and gene flow were sufficient to preserve genetic diversity despite recent forest contraction and expansion. Because beech has long distance pollen and seed dispersal, these results illustrate a ‘best case scenario’ for the maintenance of high genetic diversity and adaptive potential under climate-change-related range change.
Similar content being viewed by others


Introduction
Most species experience range expansion or contraction at some point in their history, which can have long-term consequences for population’s genetic diversity (Excoffier et al. 2009). Range change studies are of increasing interest, as we attempt to predict how climate change may induce shifts in species distributions, for example migration to higher latitude or elevation (Lenoir et al. 2008), or expansion into previously inaccessible areas (Pluess 2011). Not only do we need to predict the likelihood and direction of range shifts, we also need to understand their genetic consequences, particularly on short time scales (a few generations), as this will facilitate the development of practical guidance for conservation and sustainable management of genetic resources.
When a population is increasing in number and is also expanding spatially, several factors determine genetic diversity and structure along the colonization front (Excoffier et al. 2009). When only a few individuals contribute to the advance of the colonization wave (i.e. pulled wave following Roques et al. 2012), we expect strong repeated founder effects, increasing frequencies of few neutral mutations (“gene surfing”), loss of genetic diversity and strong spatial genetic structure (SGS) along the expansion axis (Edmonds et al. 2004; Hallatschek and Nelson 2008). Alternatively, colonization driven by many dispersing individuals (i.e. pushed wave) should result in higher genetic diversity at the colonization front and a weaker SGS, especially if these many individuals originate from a variety of locations, demes or patches. The effective number of founders depends on long-distance dispersal and on a variety of demographic processes and life-history traits determining whether the colonization wave is pushed or pulled (Hallatschek and Nelson 2008). The balance between dispersal distance relative to inter-patch distances, reproduction rate and carrying capacity is a first determinant of the pulled/pushed nature of a front (Klopfstein et al. 2006). The precise shape of the dispersal kernel has been shown to affect the effective number of founders in a complex way: roughly, fatter-tailed kernels (i.e. those that decrease more slowly at long distances) promote diversity in the front (Fayard et al. 2009) although this pattern is not completely monotonic (Paulose and Hallatschek 2020) nor scale-free (Bialozyt et al. 2006). Demographic processes, such as Allee effects (Roques et al. 2012), or life-history traits, such as a long juvenile stage (Austerlitz et al. 2000), also increase effective population size and limit the erosion of genetic diversity along the colonization front (i.e., pushed colonization waves). By extension, absence of Allee effect or short lifespan can favour the contribution of a few individuals to the colonization front (e.g., the further forward individuals, or first mature individuals), and thereby the rapid erosion of genetic diversity (i.e., pulled colonization waves).
Temperate forest trees are compelling study systems for investigating the relationship between range shifts and population genetic diversity, because experimental studies in tree species generally poorly support the expectations of classical models based on a drift-mutation model in the non-spatialized context of an isolated population or metapopulation. Classical models predict that population size reduction would be associated with decreased allelic richness and heterozygosity at neutral loci (Nei 1975). In contrast to these expectations, temperate forest trees retain high levels of within-population diversity despite their well-documented rapid post-glacial recolonization history during the last Quaternary (e.g. Petit et al. 2003; Hewitt 2004). Although decreasing trends of allelic richness along the postglacial expansion front have been reported in several tree species (e.g., Comps et al. 2001; Hoban et al. 2010), the founder events associated with postglacial range expansion have generally resulted in weak or no genetic drift. Studies of more recent and smaller scales natural expansion also generally reveal only weak genetic drift associated with founder events, and high levels of within-population diversity (Troupin et al. 2006; Born et al. 2008; Pluess 2011; Shi and Chen 2012; Lesser et al. 2013; Elleouet and Aitken 2019). Similarly, recently translocated tree populations generally combine a high level of differentiation for adaptive traits, suggesting rapid genetic evolution, with a high level of within-population diversity, indicating a limited impact of genetic drift and purifying selection (e.g. Lefevre et al. 2004). Hence, it is widely accepted that founder events can lead to genetic drift only in extremely isolated tree populations, such as described by Ledig (2000) for Pinus coulteri, where highly isolated populations are restricted to high elevations and separated by semiarid habitats which severely limit gene flow.
The theoretical expectations and empirical results described above suggest that an optimal strategy to detect the genetic signature of recent range shift in forest trees should combine several indicators, including genetic diversity and differentiation, in a spatially explicit context, including in particular SGS. Although isolation by distance (IBD) and SGS were first described by Wright (1942) in a stable metapopulation as the equilibrium resulting from geographically restricted dispersal, ongoing processes that are not at equilibrium can also be investigated by measuring the correlation between genetic divergence and geographical distance (i.e. SGS). SGS can be investigated between individuals in a continuous population (i.e., fine scale SGS) typically by using individual (dis)similarities to estimate genetic divergence, or between populations (i.e., inter-population SGS) typically by using FST to estimate genetic divergence. The conceptual frameworks of SGS and IBD apply in similar ways at these two scales of analysis (Rousset 1997, 2000). Within a recently colonized population, fine scale SGS among individuals is expected to start from no SGS, especially if the different founders are distributed randomly at arrival, and then to increase with time, especially if seed dispersal is spatially limited. It thereby provides a temporal proxy of the establishment date (Slatkin 1993; Troupin et al. 2006). For instance, Pluess (2011) found significant fine-scale SGS in late successional populations but no SGS in early successional populations. Successive founder events along a colonization axis can also lead to significant SGS among populations, thus mimicking the signature of IBD, particularly under stepwise expansion (de Lafontaine et al. 2013). In that case, though, a decrease of genetic diversity occurs jointly with the establishment of the SGS, unlike in the equilibrium IBD pattern. Only few studies compared inter-population SGS in refugia vs. expanding areas. One of these (de Lafontaine et al. 2013) found stronger genetic differentiation among populations in post-glacial refugia than in recolonized areas, but regional SGS was lower within refugia than within recolonized areas.
Here, we investigate the genetic impact of range change in the tree species Fagus sylvatica (European beech) on the slopes of Mont Ventoux, France. Across Europe in the 20th century, large areas of agricultural land were abandoned and left to secondary succession (Sluiter and De Jong 2007). In line with this pattern, the beech forests on Mont Ventoux contracted until the 19th century due to human activities, but have now recolonized areas of both the north and south slopes. In a previous study (Lander et al. 2011), we used historical records to locate most of the probable remnant populations of the massif. These beech populations hence provide a valuable model system for studying the genetic impacts of recent local population contraction and expansion, which has occurred for many plant species across Europe. We also demonstrated significant demographic fluctuations across the area using a combination of historical information and approximate Bayesian computation (ABC) analyses of modern genetic data. However, these ABC genetic analyses did not account for the spatial component of genetic structure.
In this study, we improved our spatial sampling and more deeply analysed the georeferenced genotypes to address two main questions:
(Q1) Can genetic diversity and structure provide evidence of expanding populations’ origin (i.e. natural recolonization versus establishment through planting)? Evidence of beech plantations established using both local and non-local seeds was found in historical records (Lander et al. 2011). A prerequisite to investigate the relationship between range shifts and population genetic diversity in our study system is to identify the planted populations, to avoid possibly confounding effects of plantation. Indeed, planted populations are expected to be differentiated from the others, particularly if non-local material was used. Their diversity could be higher than neighbouring populations (due to mixing of seed lots). They should also decrease the overall pattern of inter-population SGS, and show no or weak fine-scale SGS.
(Q2) Did the contraction-recolonization history reduce genetic diversity? Along the expansion front, we expect genetic diversity to decrease with increasing distance to refuges, with a potentially strong impact of the modalities of expansion: under stepwise expansion, we expect IBD patterns at landscape scale and a regular decrease in diversity with increasing distance to refuges. Alternatively, under frequent events of long-distance colonization, we expect no IBD patterns at landscape scale, and more erratic patterns of diversity with increasing distance to refuges. Within refuges, we expect higher levels of diversity, and IBD patterns at landscape scale.
Material and methods
Study species
European beech (F. sylvatica L., Fagaceae) is a common European diploid (2n = 24), monoecious tree species which typically begins to reproduce after 40–50 years. Pollen is wind-dispersed, and mating occurs almost exclusively through outcrossing, though selfing is possible (Gauzere et al. 2013). Seeds are produced in irregular mast years, and dispersed primarily by gravity, and then by various animals. Previous studies of beech on Mont Ventoux found that average dispersal distances were low for both seeds (18 m) and pollen (52 m), but both seed and pollen dispersal kernels were fat-tailed. The proportion of seeds/seedlings finding no compatible parents within plot (with typical size of 1.6 ha) was non-negligible: 46% for male parent and 11.6% for female parent on average (Gauzere et al. 2013; Bontemps et al. 2013; Oddou-Muratorio et al. 2018).
Study site and sampling design
Mont Ventoux is located at the warm and dry southern margin of the European beech distribution (Fig. S1), and the climate is typical of low altitude mountains with Mediterranean influences (weather station of Mont Serein, 1445 m a.s.l., 1993–2006; mean annual temperature of 6.8 °C and mean annual rainfall of 1300 mm). Nevertheless, the strong altitudinal variation over this large mountain, culminating at 1912 m, offers a wide array of bioclimatic conditions. The forests on the mountain have changed species composition and contracted and expanded many times due to climate cycles, however for the last 5000 years the higher elevations have been dominated by European beech and European silver fir (Barbero and Quezel 1987). Human activities caused extensive deforestation of the mountain from the 13th to the 19th centuries, and in response a reforestation programme was launched in 1861 (Jean 2008). In a previous study (Lander et al. 2011), we used historical data to identify a modern population minimum in 1845, and we were able to distinguish areas of beech forest, which have been present for the last 200 years (four refugia) from two areas that appear to be the result of recent forest expansion (Fig. 1). That analysis found that the three regions under study (North-West, North-East, South) were genetically distinct, with two remnant areas (North-West, North-East) and one area of recent expansion (South). However, the areas of recent expansion, as well as the remnant population on the South ridge were under-represented in that previous study, which used 1932 trees in 51 plots. Moreover, the previous study did not explicitly account for historical records showing that beech was planted on the South edge of Mt Ventoux (Lander et al. 2011), although much less intensively than other species (e.g., Pinus nigra, Pinus sylvestris). These beech plantations reportedly used either local seeds (raised in local, non-permanent, “flying” nurseries) or non-local seed delivered by commercial nurseries.

Historical records allowed us to distinguish four refugia area (filled polygons), where beech has been present for the last 200 years (NW_REF and NE_REF on the Northern slope; SW-REF and SE-REF on the Southern slope) from two area of recent expansion (hatched polygons; S-EXP and NE-EXP). The spatial delimitation between the NW_REF and NE_REF was chosen to be a large terrace, while that between S-EXP and NE-EXP was chosen to be the major crest line. SW-REF and SE-REF were aggregated in most analyses as only few plots could be sampled in these areas. Plots are mapped with shape indicating the region (filled dots = NW_REF; filled squares = NE_REF; empty squares = NE_EXP; filled triangles = S_REF; empty triangles = S_EXP).
For this new study, we sampled 600 additional trees in 20 new plots, providing a total of 2532 adult trees in 71 plots covering five different regions of Mont Ventoux (Fig. 1). We retained 2042 trees for analyses (see Appendix A1 for selection), distributed as follows: (1) 748 trees in 25 plots in the north-western refuge (NW_REF), which is a tight mixture of remnant and more recent communal forest under traditional management; (2) 464 trees in 16 plots in the north-eastern refuge (NE_REF) which is included in a Biosphere Reserve and is unmanaged; (3) 316 trees in 12 plots in the far eastern region, recently recolonized by European beech expanding out of the refuge areas (NE_EXP); (4) 208 beech in 7 plots at high elevation on the south face of Mont Ventoux, a region identified as refuge forest (S_REF); and (5) 306 beech in 11 plots at low elevation on the south face of Mont Ventoux, a region recently recolonized by European beech (S_EXP).
Within each plot, 28.8 adult trees on average (up to a maximum of 40 individuals) were sampled in an area of ~50 m radius so that all trees were separated by at least 3 m. All trees had a circumference at breast height >160 mm. Trees were chosen so that half of them had the largest circumference in the plot (“Old” trees, average mean/maximal circumference = 958/1495 mm) and the other half had the smallest circumference (“Young” trees, average mean/minimal circumference = 444/309 mm). Geographical coordinates were recorded for all sampled trees and a map of the study area was developed in ArcMap 10.4 (ESRI) using the geographical coordinates of the trees, a map of current forest ownership and forest cover (Office National des Forêts 2001), and a topographical map (IGN-PACA 2002). Plots’ altitudes were estimated in ArcMap. Finally, the maximal age of a tree within each plot was estimated based on the tree ring profile of the largest possible tree (average maximal age = 155). Detailed information per region and plot is available in Tables 1, S1, Fig. S2 and Appendix A1.
Genotyping and basic statistics
All individuals were genotyped using 13 microsatellite markers, one of which was excluded due to high frequency of null alleles. Detailed information on genotyping and quality of the marker set is available in Supplementary Appendix A1.
Statistical analyses were conducted using R 3.6.2 (R Core Team 2019) unless otherwise indicated. We considered several statistics to describe population diversity at plot level. We first used the package ‘diveRsity’ (Keenan et al. 2013) to compute the allelic richness (Ar) and the expected heterozygosity (He). We also computed He and Ar values for each cohort within plot, and derived the difference in He and Ar between old and young individuals (respectively difHe and difAr). We used the package ‘hierfstat’ to compute Wright’s inbreeding coefficient (FIS), pairwise FST among plots following Weir and Cockerham (1984), and βWT, a plot-specific index of genetic differentiation relative to the entire pool (Weir and Goudet 2017). Tests for departures from Hardy–Weinberg equilibrium and linkage equilibrium were conducted using Fstat 2.9 (Goudet 1995). We used the package ‘hierfstat’ to estimate the components of variance in allelic frequencies among regions, among plots within regions, and among individuals within plots, and derived the associated F-statistics (FCT, FSC, FIS and FST).
Bayesian inference of population structure
The genetic structure was investigated using two different tools based on Bayesian clustering algorithms. These methods have different prior distributions and assumptions, and we used them simultaneously to evaluate the robustness of the genetic clusters. We hypothesized that the number of possible clusters (K-values) was unlikely to be >4 in our case, considering the continuous, rather than patchy, distribution of beech on Mont Ventoux, and the presence of only four beech refugia on the mountain during the modern population minimum. However, because of the possible planting using non-local seeds, we investigated a wider range of K-values.
Bayesian clustering of the genetic data was first performed using STRUCTURE 2.3.3 (Pritchard et al. 2000), with K varying between 1 and 13, and 10 runs for each K value. Parameters were 2500 burn-in periods and 10,000 Markov Chain Monte Carlo repetitions after burn-in, with allele frequencies correlated among populations and an admixture model of population structure. To account for non-independence between two genotypes from the same population, we used population identifiers as prior information to assist clustering. The ΔK statistics allowed us to evaluate the change in likelihood and select the optimal K value (Evanno et al. 2005). For the selected K-value, we averaged over 10 runs the proportion of each cluster in each sampling plot and the individual probabilities of belonging to each cluster using CLUMPAK (Kopelman et al. 2015).
TESS 2.3 (Chen et al. 2007) was also used to estimate the number of genetic clusters present in the data by incorporating the geographical coordinates of individuals as prior information to detect discontinuities in allele frequencies. We used an admixture model and a burn-in of 10,000 iterations followed by 50,000 iterations from which estimates were obtained. We performed 200 independent runs for each K value (K = 2–6), with spatial interaction influence ψ at 0.6 (default value). The optimal K value was determined by the lowest value of the deviance information criterion (DIC). The 200 runs for the best K were averaged using CLUMPP (Jakobsson and Rosenberg 2007).
Spatial outputs of both STRUCTURE and TESS were visualized using the R script ‘krigAdmixProportions’ distributed with the TESS programme. This script uses a kriging approach to interpolate a surface model based on scattered, spatially explicit, data points. This consists in using the proportions of the different clusters at each of the 71 sampled locations to estimate the probabilities to belong to the different clusters at all locations of the landscape.
Spatial variation in diversity and connectivity
We visualized spatial patterns in genetic diversity and geneflow rates using the programme estimated effective migration surfaces (EEMS; Petkova et al. 2015). This method uses sampling localities and pairwise dissimilarity matrices calculated from microsatellite data to identify regions where genetic similarity decays more quickly than expected under IBD. A user‐selected number of demes determines the geographic grid size and resulting set of migration routes, and the expected dissimilarity between two samples is approximated using resistance distance. These estimates are calculated without the need to include environmental variables or topographic information and are subsequently interpolated across the geographic space to provide a visual summary of observed genetic dissimilarities, including regions with higher and lower gene flows than expected. We tested three numbers of demes (400, 600, 800) using the runeems_sats version of EEMS. For each deme size, we ran three independent analyses, with a burn‐in of 500,000 and MCMC length of 3,000,000. The results were combined across the three independent analyses, and convergence of runs was assessed using the ‘reemsplots’ R package. Using this package, we generated surfaces of effective diversity (q) and effective migration rates (m) combining the nine independent runs for the three deme size.
Isolation by distance
We first estimated SGS among sampling plots across the whole study area and tested whether geographic distances significantly shaped the patterns of genetic differentiation, estimated by FST, among plots using the software SpaGeDi 1.4c (Hardy and Vekemans 2002). To test IBD, the FST values were regressed on ln(dij), where dij is 3D spatial distance accounting for elevation between plots i and j, calculated using the 3D Analyst Tools in ArcMap 10.4. Then, we tested the regression slope (blogFST) using 5000 permutations of genotypes among population’s positions. These analyses were run globally over the 71 plots, and within each region. SGS estimates can be sensitive to outlier plots showing higher or lower differentiation for the others plots (de Lafontaine et al. 2013). To account for possible biases due to planted forest material, we ran conservative SGS analyses within each historical group (see “Results” section).
We also estimated fine-scale SGS within each plot with SpaGeDi. Within each plot, genetic relatedness between all pairs of individuals i and j was estimated using the kinship coefficient Fij (Loiselle et al. 1995). To estimate SGS, the Fij values were regressed on ln(dij), where dij is the 2D spatial distance between individuals i and j. We tested the significance of SGS (regression slope, blogFij) using 5000 permutations of genotypes among individual positions. Following Vekemans and Hardy (2004), the SGS intensity was quantified by Sp = blogFij/(F1–1), where F1 is the average kinship coefficient between individuals of the first distance class (<10 m). Sp primarily depends upon the rate of decrease of pairwise kinship coefficients between individuals with the logarithm of the distance, and is scaled by the average level of relatedness between individuals, which allows inter-population comparison.
Impact of recolonization history on genetic diversity
We tested the hypothesis that the distance and steepness of up-slope and down-slope travel between each study population and the ‘core area’ of each of the refugia (NW_REF, NE_REF, SW_REF and SE_REF) affects genetic diversity. As the refugia are irregularly shaped, the ‘core areas’ were defined as the medial axes of the refugia polygons (‘Thin’, ArcMap 10.4). Following Zafar (2011), the 2D line from the centroid of each sample plot to the nearest point on the medial axis of each of the four refugia was drawn using Analysis Tools (ArcMap 10.4; Supplementary Table S1). The 2D lines were then converted to 3D lines based on two aspect rasters, one weighted for travel north to south, and one for travel south to north (3D Analyst, ArcMap 10.4), providing data on the linear distance and travel up and down a seed or seed vector would have had to travel on Mt Ventoux’s surface between each refuge and each study population.
Similar to Hoban et al. (2010), we used ANCOVA to investigate how the distance to refugia shaped genetic variation at plot level, described by seven summary statistics (He, Ar, FIS, Sp, βWT, difHe, and difAr), considered as response variables. For each summary statistic, we considered the following models:
Response variable = wdistNE + wdistNW + wdistSE + wdistSW + HistGroup (model 1)
Response variable = (wdistNE + wdistNW + wdistSE + wdistSW)×HistGroup (model 2)
where all distances are quantitative variables, and HistGroup is a categorical variable integrating the recent history of plots as supported by genetic clustering analyses (i.e., refuge, expansion area, and planted plots; and see “Results” section). The best linear model was selected based on AIC with the stepwise algorithm implemented in the step procedure of the ‘stats’ package.
Results
Genetic variation within and among sampled plots
Genetic diversity estimates are summarized in Table 1 and detailed in Table S2. In total, 154 alleles were scored at the 12 loci, corresponding to an average of 12.8 alleles per locus (range = 5–23). Mean allelic richness per population ranged between 3.1 and 5.11 (mean Ar = 4.3), while observed and expected heterozygosities per population ranged from 0.600 and 0.610–0.760 and 0.720 (mean Ho = 0.683 and mean He = 0.667). Ar, He, and Ho did not differ significantly among the five studied regions.
Ten populations showed significant departure from Hardy–Weinberg equilibrium, four of which displayed heterozygosity deficit and another six showed heterozygosity excess. FIS-values ranged from −0.096 to 0.108 (mean FIS = 0), and did not differ between regions. Genetic differentiation of each plot from the entire population ranged from −0.027 to 0.11 (mean βWT = 0.03). The region NW_REF showed a significantly lower βWT-value, likely due to the high contribution of the 25 plots of this region to the entire genepool.
Estimation of hierarchical variance components showed that most of the genetic variation lies among individuals within plots (Table S3): genetic differentiation among-plots within regions (FSC = 0.029), and among-regions (FCT = 0.002) were weak, although significant. The overall genetic differentiation among plots was FST = 0.031.
Among-plots pairwise FST-values ranged between 0 and 0.097 with a mean value of 0.031 (median = 0.029) (Fig. S3). The highest observed differentiation values involved plots E_1231, S_1913 and S_2007. The lowest observed differentiation values occurred between plots of the South region
Spatially distinct genetic clustering
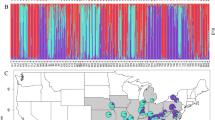
The genetic clustering analyses found weak but significant genetic structure. Using STRUCTURE, the method of Evanno et al. (2005) selected K = 3 and K = 6 as the most-likely values of the number of clusters (Fig. S4a). Retaining K = 3 as the first major peak in ΔK statistics, spatial kriging of the Q-matrix suggests that the three clusters are spatially distinct (Figs. 2 and S4b–e). Cluster C1 is predominant in North-West (22 of 25 plots) and North-East (13 of 16 plots) refuges. Cluster C2 is predominant in South refuge (4 of 7 plots), and present in all other regions. Cluster C3 groups plots S_1727, S_1913, and S_3 (South Expansion), plots E_1231 and E_1755 (East expansion) and plot S_23 (on the southern boundary of the SE refuge). Finally, the average allelic divergence (FST) between clusters C1 and C2 is ~1.3%, while FST between clusters C3 and C1 (C2 respectively) is 2.3% (2.6% respectively). When using STRUCTURE without prior information, the power of plots’ assignation to clusters C1 and C2 decreased, while cluster C3 remained distinct (Fig. S5).

The colours red coral, ochre and aquamarine correspond, respectively, to clusters C1, C2 and C3. The colour intensity indicates the probability to belong to the dominant cluster at a given position in space, based on spatial kriging of the individual q-matrix (see Fig S4 for additional information). Plots shape indicates the region (see legend of Fig. 1). Grey lines represent topographic isoclines.
In the TESS analysis the lowest DIC value was for K = 6 (Fig. S6). For K = 3 (Fig. S6), TESS clustering is fully consistent with STRUCTURE, as illustrated by the strong correlations between the membership coefficients of plots to clusters estimated with TESS and STRUCTURE (ρ = 0.96 for C1 and C2; ρ = 0.99 for C3, p-values < 0.001). Cluster C3 is also the most supported: it appears when the results from K = 2 are graphed, and remains distinct up to K = 6 (Fig. S6).
In the following IBD and historical analyses, to test different expectations for expansion areas vs. refuges, we accounted for the detected genetic clusters and classified the 71 plots in three historical groups. The “refugia” group (REF) includes 48 plots from the refuge regions (i.e. 25 NW_REF plots, 16 NE_REF plots and 7 S_REF plots), all assigned either to clusters C1 and C2. The “likely planted” group (“PLANTED”) includes four S-EXP plots and two NE-EXP plots predominantly assigned to cluster C3 (six plots in total). The “natural expansion” group (EXP) includes all of the remaining 17 plots of the expansion area (i.e. 10 NE-EXP and 7 S-EXP plots), assigned to clusters C1 and C2 (Table S2).
Spatial differences in geneflow and genetic diversity
EEMS spatial analyses highlight several barriers to migration resulting from either historical or contemporary patterns of gene flow (Figs. 3 and S7). There is evidence for restricted migration around the mountaintop, and along the expansion paths towards the East and South. Spatial analyses of genetic diversity highlight four main regions of exceptionally high diversity, three of which are located along the expansion paths towards the East and South, and the last one in NW refuge. However, regions with lower‐than‐expected genetic diversity are also found along the expansion paths towards the East and South, resulting in a tight mosaic of diverse and homogenous areas in term of genetic composition.

a Contour maps representing the posterior mean of effective migration surface, where blue colours represent areas of high migration, or dispersal corridors, whereas orange regions represent areas of low migration, or dispersal barriers. b Contour maps representing the posterior mean of effective diversity surface, where orange regions indicates areas of lower‐than‐expected genetic diversity, and blue colours represent higher levels of genetic diversity. The light grey dots illustrate the sampling design (bigger dots indicating a deme with more samples). Grey lines represent topographic isoclines.
Isolation by distance
SpaGeDi found a significant signal of IBD on genetic differentiation between the 71 plots (Table 2, Fig. 4). Pairwise FST overall significantly increased with increasing 3D geographic distances accounting for elevation (blog3D = 0.003, p-value < 0.001). However, this significant pattern of IBD is mainly driven by the 25 plots of the NW refuge (blog3D = 0.007, p-value < 0.001), while no significant IBD patterns were detected in other regions. The signature of IBD remained significant between the 48 plots from the refugia group (REF), although weaker than that of the NW refuge (blog3D-REF = 0.0023 versus blog3D-NW_REF = 0.007). No signature of IBD could be detected between the 17 plots from the expansion group (EXP), or between the six plots of the likely planted group (PLANTED), which may be due to weak testing power.

Solid (respectively, broken) lines indicates region where SGS is significant (respectively, not significant). Filled (respectively, hatched) symbols represent average FST values lower (respectively, higher) than expected under complete spatial randomness. Shapes and colours indicate a the region (green dots: NW_REF; blue squares: NE_REF; light blue squares: NE_EXP; red triangles: S_REF; orange triangles: S_EXP); b the historical group (purple dots: refugia; orange squares: natural expansion; aquamarine triangle: likely planted).
A significant signal of IBD on kinship coefficients among individuals within plot (i.e., fine-scale SGS) was detected in 37 of the 71 plots, corresponding to 17% of the NE_EXP, 27% of the S_EXP, 50% of the NE_REF, 72% of the NW_REF and 86% of the S_REF (Table S2). Although the prevalence of SGS was higher in refugia than in expansion areas (χ2 test p-value = 0.003), the intensity of SGS, as depicted by Sp, did not significantly differ among regions.
Impact of recolonization history on genetic diversity
ANCOVA analyses showed that the impacts of distance to the refugia on genetic diversity at plot level varied depending on the summary statistics considered (Table 3, Fig. 5). We found that Nei’s genetic diversity (He) significantly decreased with increasing distance to the NW refuge in the “REF” and “PLANTED” groups, while He significantly increased with increasing distance to the NW refuge in the “EXP” group. We detected significantly higher allelic richness (Ar) in the “PLANTED” group as compared to the “REF” and “EXP” groups, but no significant effect of the distance to refuge on Ar.

a Variation of expected heterozygosity (He) with the distance to the NW refuge, b variation of allelic richness (Ar) among the historical groups, c variation of differentiation from the entire gene pool (βWT) with the distance to the NW refuge and d variation of inbreeding coefficient (FIS) among the historical groups.
Regarding Wright’s inbreeding coefficient (FIS), without accounting for distances to the refuges, we found a significantly higher FIS-level in the “PLANTED” group compared to the “REF” and “EXP” groups (Fig. 5d). Moreover, FIS overall significantly increased with increasing distance to the NE and NW refuge (which is partially confounded with the “PLANTED” origin). The genetic differentiation relative to the entire pool (βWT) increased significantly with increasing distance to the NW refuge in the “REF” and “PLANTED” groups, while βWT significantly decreased with increasing distance to the NW refuge in the “EXP” group (Fig. 5c).
Fine-scale SGS was significant in 66% of the “REF” plots, in 29% of the “EXP” plots, and in none of the “PLANTED” plots (χ² test p-value = 0.0008). However, the Sp statistics did not reveal significant pattern variation in the intensity of SGS among groups, except a marginally significant trend for lower SGS intensity in the “PLANTED” group as compared to the “REF” and “EXP” groups (p-value = 0.08). Finally, increasing distance to NE refuge and decreasing distance to SW refuge were associated with decreasing difference in Ar between old and young individuals (difAr), while no significant pattern was observed for difHe.
Discussion
This study aimed at investigating the genetic consequences of recent range shift, using a spatially explicit theoretical framework and a valuable study system, that of the recent expansion of beech on Mont Ventoux. We first discuss how the possible establishment of some plots through planting may interfere with the signature of natural recolonization. We then summarize how the observed patterns of genetic diversity and structure, including SGS, support the theoretical expectations on the genetic consequences of spatial population expansion. Finally, we discuss how these findings can be used to guide the management of beech populations.
Genetic signatures of population origin
Beech has been a dominant species for 5000 years on Mont Ventoux, although its spatial range has contracted and expanded several times, partly due to human activities in the last 1000 years (Lander et al. 2011).
Our results provide genetic evidence of tree planting events in six plots of the South and North-East expansion areas. These plots all cluster together with Bayesian structure analyses, and on the FST-based Neighbour-Joining tree (Fig. S3). Moreover, they have a significantly higher inbreeding coefficient (possibly due to Wahlund effect) and higher levels of allelic richness, two features consistent with the mixing of seed lots from different origins. Only two plots among the six identified are over-differentiated from all other plots (up to pairwise FST = 0.09, as compared to the mean FST = 0.03), suggesting that the planted material was most often of local origin. Interestingly, the EEMS analyses tend to associate these six plots with areas combining low geneflow connectivity and high diversity, a paradox also consistent with planting rather than natural establishment. Finally, none of these six plots shows significant SGS at the individual level.
Thus six plots of the 23 sampled in the expansion area (26 %) appear to originate from planting during the reforestation programme launched in 1861. Hence, we cannot exclude the possibility that planting by humans contributed to the spread rate previously estimated for beech on Mont Ventoux (27–38 m/year, Lander et al. 2011). However, the remaining 17 expansion plots (73%) seem to have established naturally, confirming the high ability of beech to spread and colonize new areas. To test theoretical hypothesis on the genetic consequences of natural population expansion, we thus carefully distinguished the six probably planted plots from the 17 plots probably originating from natural regeneration.
A weak but significant impact of contraction/expansion history on genetic diversity
Despite the short time elapsed to allow pollen to wipe out founder effects due to seed dispersal, average levels of genetic diversity and structure did not show much evidence of the impact of recent local range shifts. In particular, plots in expanding areas did not reveal the classical signatures of strong genetic drift associated with founder events: their genetic differentiation was overall low (although a few plots were over-differentiated from the others), and their heterozygosity and allelic richness did not differ from the refuge areas. We cannot exclude the possibility that the refugia themselves were subject to population size contraction. However, consistent with previous studies in forest trees, our results confirm that the long juvenile phase and the predominance of high pollen flow in wind-pollinated trees strongly attenuate the genetic impacts of demographic changes (Shi and Chen 2012; Lesser et al. 2013; Elleouet and Aitken 2019).
On the other hand, spatial patterns of genetic diversity and structure did reveal the imprint of the expansion process, with a signature typical of genetic mixing between refuges. Indeed, heterozygosity increased with increasing distance from the Northwest refuge, while genetic differentiation from the entire gene pool (βWT) decreased. These findings are consistent with the increasing heterozygosity along post-glacial colonization axes at European scale reported by Comps et al. (2001). Such an increase in He and decrease in βWT is likely to result from the combination of several processes and life history traits. First, a very fat-tailed pollen dispersal curve as estimated for beech (Gauzere et al. 2013) can result in mixing of propagules from different distant sources (Klein et al. 2006) and can thus promote high genetic diversity at the colonization front (e.g. Fayard et al. 2009; Paulose and Hallatschek 2020). Second, the long juvenile phase strongly attenuates founder effects during colonization in a diffusive dispersal model (Austerlitz et al. 2000). Third, admixture at the intersection of colonization fronts from different refugia can increase diversity (Comps et al. 2001), even though this effect may be limited in our study case with weak genetic differentiation among local refugia. Finally, selection pressures in the open environmental conditions at the colonization front may support the maintenance of heterozygosity (Comps et al. 2001).
Another spatial signature of recent expansion was the absence of inter-population IBD, whereas significant inter-population IBD was detected in some refuge areas. Moreover, fine-scale SGS was much more prevalent in refuge as compared to expansion areas. This is consistent with the theoretical work of Slatkin (1993) on IBD, which showed that a species having restricted dispersal should exhibit SGS if enough time has elapsed after establishment, assuming no initial structuring (Troupin et al. 2006). Testing this hypothesis in beech, de Lafontaine et al. (2013) showed the reverse pattern, where recently colonized populations displayed significant SGS whereas more ancient populations did not. However, they focused on post-glacial recolonization, where several tens of generation probably allowed SGS to develop, unlike our case of a recent colonization. Our study also highlights the idea that fine-scale SGS does not systematically occur in refugia, for instance in the North East and South refugia, which is also typical of a mix of founder origins under very fat-tailed kernels (Paulose and Hallatschek 2020).
Overall, this study confirms our previous results (Lander et al. 2011), and shows that the genetic signatures of expansion predominate over those of genetic drift related to founder events. The previous ABC approach selected the scenario were three populations (NorthWest, NorthEast, South) expanded from a smaller ancestral population, rather than a scenario with bottleneck supported by the historical data. This study additionally highlights specific spatial signatures of the expansion process. Moreover, the South population was the most divergent with the previous ABC approach. Here, we showed that two plots of this South population (which included only five plots) originate from planting rather than natural establishment, shedding light on this higher divergence.
Consequences for the management of beech populations
Knowledge of colonization and dispersal processes is crucial for management planning and conservation efforts, particularly with a view to managing invasive species or genotypes (Brandes et al. 2019), or to predicting species’ response to climate change (Jump and Peñuelas 2005). However, empirical tests of how range expansion or contraction shape levels of diversity within and among populations are limited by our ability to collect data on an appropriate spatial and temporal scales, particularly in long-lived species. Many studies investigate these issues at large spatial and temporal scales, which provide useful insights for the conservation and management of genetic resources at species distribution scales. However, local management also requires studies investigating recent and rapid events of range change, similar to those expected under ongoing global and climate changes.
Bioclimatic niche models predict a future reduction of beech at the rear edge of its range over the next few decades (Cheaib et al. 2012; Dyderski et al. 2018). Moreover, forest areas across Europe, and in the Mediterranean basin in particular, contracted and expanded many times in the last centuries following socio-economic changes and their consequences on land-use patterns (Sluiter and De Jong 2007). Beech forests were no exception, and large areas of beech forest have been cleared for agricultural production, and then recolonized following field abandonment. This study found evidence of high adaptive potential of beech despite such local range changes. First, our results demonstrate an overall increase in heterozygosity and decrease in genetic differentiation along the expansion front, consistent with the genetic mixing of founders from different origins. A major reservoir of genetic diversity was identified within one refugia area (NorthWest). Moreover, the overall high levels of genetic diversity and low genetic differentiation over the 15,000 ha study area confirms the capacity for extensive gene flow and the large effective population size previously estimated (Lander et al. 2011; Oddou-Muratorio et al. 2018). These high levels of gene flow can be expected to foster rather than hamper local adaptation, as shown by the microgeographic adaptation patterns along the northern altitudinal gradient reported by Gauzere et al. (2020). Finally, this study also demonstrates that the levels of allelic richness and heterozygosity are stable over time between old and young cohorts (−0.6% and −0.1%, respectively, on average). Some reduction up to 20% in He or Ar could be detected locally, but not associated with the expansion process, or with differences in management practises (e.g., traditional management in NorthWest refuge versus no management in the NorthEast refuge).
In conclusion, this study showed that range change on a local scale and over a small number of generations did leave detectable genetic signatures, but overall did not increase genetic differentiation, nor reduce heterozygosity or allelic richness. These results paint a positive picture of the potential for species to maintain genetic diversity and adaptive potential through climate-change-related range change. However, beech is both wind-pollinated and biotically dispersed, and therefore expected to have long distance pollen and seed dispersal; moreover, the seeds in this study would mainly have been dispersing downhill on the massif; finally, the local persistence of several refugia and additional scattered beech trees even at the population minimum is likely to have allowed continued geneflow between populations to be maintained. For these different reasons, our results represent a ‘best case scenario’ for the maintenance of high genetic diversity at the population perimeter during population expansion, including during climate-change-related range change. Other tree species with more limited dispersal abilities, and/or more scattered distribution, such as P. coulteri (Ledig 2000), would represent an alternative ‘worse case scenario’. The results therefore should be conservatively interpreted as they relate to the development of management recommendations for forest trees, and in particular for the high conservation priority forests of the Mediterranean Basin (Madon and Médail 1997; Médail and Diadema 2009).
Data archiving
Data are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.nvx0k6dqt
References
Austerlitz F, Machon N, Gouyon P, Godelle B (2000) Effects of colonization processes on genetic diversity: Differences between annual plants and tree species. Genetics 154:1309–1321
Barbero M, Quezel P (1987) La végétation du Ventoux: diversité, stabilité et utilisation actuelles des écosystèmes. Etud Vauclus 3:79–84
R Core Team (2019) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Bialozyt R, Ziegenhagen B, Petit RJ (2006) Contrasting effects of long distance seed dispersal on genetic diversity during range expansion. J Evol Biol 19:12–20
Bontemps A, Klein EK, Oddou-Muratorio S (2013) Shift of spatial patterns during early recruitment in Fagus sylvatica: evidence from seed dispersal estimates based on genotypic data. Ecol Manag 305:67–76
Born C, Kjellberg F, Chevallier MH, Vignes H, Dikangadissi JT, Sanguié J et al. (2008) Colonization processes and the maintenance of genetic diversity: Insights from a pioneer rainforest tree, Aucoumea klaineana. Proc R Soc B 275:2171–2179
Brandes U, Furevik BB, Nielsen LR, Kjær ED, Rosef L, Fjellheim S (2019) Introduction history and population genetics of intracontinental scotch broom (Cytisus scoparius) invasion (A Zhan, Ed.). Divers Distrib 25:1773–1786
Cheaib A, Badeau V, Boe J, Chuine I, Delire C, Dufrêne E et al. (2012) Climate change impacts on tree ranges: model intercomparison facilitates understanding and quantification of uncertainty. Ecol Lett 15:533–544
Chen C, Durand E, Forbes F, François O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7:747–756
Comps B, Gömöry D, Letouzey J, Thiébaut B, Petit RJ (2001) Diverging trends between heterozygosity and allelic richness during postglacial colonization in the European beech. Genetics 157:389–397
Dyderski MK, Paź S, Frelich LE, Jagodziński AM (2018) How much does climate change threaten European forest tree species distributions? Glob Chang Biol 24:1150–1163
Edmonds CA, Lillie AS, Cavalli-Sforza LL (2004) Mutations arising in the wave front of an expanding population. Proc Natl Acad Sci USA 101:975–979
Elleouet JS, Aitken SN (2019) Long-distance pollen dispersal during recent colonization favors a rapid but partial recovery of genetic diversity in Picea sitchensis. New Phytol 222:1088–1100
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501
Fayard J, Klein EK, Lefèvre F (2009) Long distance dispersal and the fate of a gene from the colonization front. J Evol Biol 22:2171–2182
Gauzere J, Klein EK, Oddou-Muratorio S (2013) Ecological determinants of mating system within and between three Fagus sylvatica populations along an elevational gradient. Mol Ecol 22:5001–5015
Gauzere J, Klein EK, Brendel O, Davi H, Oddou-Muratorio S (2020) Microgeographic adaptation and the effect of pollen flow on the adaptive potential of a temperate tree species. New Phytol. 227:641–653
Goudet J (1995) FSTAT (Version 1.2): A computer program to calculate F-Statistics. J Hered 86:485–486
Hallatschek O, Nelson DR (2008) Gene surfing in expanding populations. Theor Popul Biol 73:158–170
Hardy OJ, Vekemans X (2002) Spagedi: a versatile computer program to analyse spatial. Mol Ecol Notes 2:618–620
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc B 359:183–195
Hoban SM, Borkowski DS, Brosi SL, McCleary TS, Thompson LM, McLachlan JS et al. (2010) Range-wide distribution of genetic diversity in the North American tree Juglans cinerea: a product of range shifts, not ecological marginality or recent population decline. Mol Ecol 19:4876–4891
IGN-PACA (2002) BDALTI_MNT - V2001, Topographical map of France 1/ 25 000 to 1/ 50 000. BD ALTI ® - © IGN PFAR 2000
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jean H (2008) Reboisement du Ventoux et restauration des terrains en montagne dans la vallée du Toulourenc. Les Carnets du Vent 61:72–75
Jump AS, Peñuelas J (2005) Running to stand still: adaptation and the response of plants to rapid climate change. Ecol Lett 8:1010–1020
Keenan K, Mcginnity P, Cross TF, Crozier WW, Prodöhl PA (2013) DiveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788
Klein EK, Lavigne C, Gouyon PH (2006) Mixing of propagules from discrete sources at long distance: comparing a dispersal tail to an exponential. BMC Ecol 6:25
Klopfstein S, Currat M, Excoffier L (2006) The fate of mutations surfing on the wave of a range expansion. Mol Biol Evol 23:482–490
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
de Lafontaine G, Ducousso A, Lefèvre S, Magnanou E, Petit RJ (2013) Stronger spatial genetic structure in recolonized areas than in refugia in the European beech. Mol Ecol 22:4397–4412
Lander TA, Oddou-Muratorio S, Prouillet-Leplat H, Klein EK (2011) Reconstruction of a beech population bottleneck using archival demographic information and Bayesian analysis of genetic data. Mol Ecol 20:5182–5196
Ledig FT (2000) Founder effects and the genetic structure of Coulter pine. J Hered 91:307–315
Lefevre F, Fady B, Ghosn D, Bariteau M (2004) Impact of founder population, drift and selection on the genetic diversity of a recently translocated tree population. Heredity 94:542–550
Lenoir J, Gégout JC, Marquet PA, De Ruffray P, Brisse H (2008) A significant upward shift in plant species optimum elevation during the 20th century. Science (80-) 320:1768–1771
Lesser MR, Parchman TL, Jackson ST (2013) Development of genetic diversity, differentiation and structure over 500 years in four ponderosa pine populations. Mol Ecol 22:2640–2652
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Madon O, Médail F (1997) The ecological significance of annuals on a Mediterranean grassland (Mt Ventoux, France). Plant Ecol 129:189–199
Médail F, Diadema K (2009) Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J Biogeogr 36:1333–1345
Nei M (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Oddou-Muratorio S, Gauzere J, Bontemps A, Rey JF, Klein EK (2018) Tree, sex and size: ecological determinants of male vs. female fecundity in three Fagus sylvatica stands. Mol Ecol 27:3131–3145
Office National des Forêts (2001) Carte des unités d’analyse pour les forêts publiques du Mont-Ventoux (ed. Direction Territoriale Méditerranée)
Paulose J, Hallatschek O (2020) The impact of long-range dispersal on gene surfing. Proc Natl Acad Sci USA 117:7584–7593
Petit RJ, Aguinagalde I, De Beaulieu JL, Bittkau C, Brewer S, Cheddadi R et al. (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science (80-) 300:1563–1565
Petkova D, Novembre J, Stephens M (2015) Visualizing spatial population structure with estimated effective migration surfaces. Nat Genet 48:94–100
Pluess AR (2011) Pursuing glacier retreat: genetic structure of a rapidly expanding Larix decidua population. Mol Ecol 20:473–485
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Roques L, Garnier J, Hamel F, Klein EK (2012) Allee effect promotes diversity in traveling waves of colonization. Proc Natl Acad Sci USA 109:8828–8833
Rousset F (1997) Genetic differentiation and estimation of gene flow from FStatistics under isolation by distance. Genetics 145:1219–1228
Rousset F (2000) Genetic differentiation between individuals. J Evol Biol 13:58–62
Shi MM, Chen XY (2012) Leading-edge populations do not show low genetic diversity or high differentiation in a wind-pollinated tree. Popul Ecol 54:591–600
Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47:264–279
Sluiter R, De Jong SM (2007) Spatial patterns of Mediterranean land abandonment and related land cover transitions. Landsc Ecol 22:559–576
Troupin D, Nathan R, Vendramin GG (2006) Analysis of spatial genetic structure in an expanding Pinus halepensis population reveals development of fine-scale genetic clustering over time. Mol Ecol 15:3617–3630
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13:921–935
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Weir BS, Goudet J (2017) A unified characterization of population structure and relatedness. Genetics 206:2085–2103
Wright S (1942) Isolation by distance. Genetics 80:1514–1520
Zafar SD (2011) Determination of an optimal bike path based on slope & distance data. Web site 17.
Acknowledgements
Historical and ecological information on sampling plots was provided by P. Dreyfus & Fl. Jean (INRAE-URFM) and J Terracole & E Jensel (ONF). We thank N. Turion, O. Gilg, F. Rei, M. Pringarbe (INRAE-UEFM) and E. Jensel (ONF) for field work, M. Correard for assistance with GIS, and M. Lingrand for genotyping and managing the lab work. This study was funded by the French Agence Nationale pour la Recherche (ANR ColonSGS; grant ANR-07-JCJC-0117), the EC-supported Network of Excellence Evoltree (GOCE-016322) and the French programme ECOGER.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lander, T.A., Klein, E.K., Roig, A. et al. Weak founder effects but significant spatial genetic imprint of recent contraction and expansion of European beech populations. Heredity 126, 491–504 (2021). https://doi.org/10.1038/s41437-020-00387-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-00387-5
This article is cited by
-
A novel synthesis of two decades of microsatellite studies on European beech reveals decreasing genetic diversity from glacial refugia
Tree Genetics & Genomes (2023)
-
A set of nuclear SNP loci derived from single sample double digest RAD and from pool sequencing for large-scale genetic studies in the European beech Fagus sylvatica
Conservation Genetics Resources (2022)
-
Coupling fossil records and traditional discrimination metrics to test how genetic information improves species distribution models of the European beech Fagus sylvatica
European Journal of Forest Research (2022)
-
Trunk perimeter correlates with genetic bottleneck intensity and the level of genetic diversity in populations of Taxus baccata L
Annals of Forest Science (2021)
