
Abstract
Isthmian Central America (ICA) is one of the most biodiverse regions in the world, hosting an exceptionally high number of species per unit area. ICA was formed <25 million years ago and, consequently, its biotic assemblage is relatively young and derived from both colonization and in situ diversification. Despite intensive taxonomic work on the local fauna, the potential forces driving genetic divergences and ultimately speciation in ICA remain poorly studied. Here, we used a landscape genetics approach to test whether isolation by distance, topography, habitat suitability, or environment drive the genetic diversity of the regional frog assemblage. To this end, we combined data on landscape features and mitochondrial DNA sequence variation for nine codistributed amphibian species with disparate life histories. In five species, we found that at least one of the factors tested explained patterns of genetic divergence. However, rather than finding a general pattern, our results revealed idiosyncratic responses to historical and ecological processes, indicating that intrinsic life-history characteristics may determine the effect of different drivers of isolation on genetic divergence in ICA. Our work also suggests that the convergence of several factors promoting isolation among populations over a heterogeneous landscape might maximize genetic differentiation, despite short geographical distances. In conclusion, abiotic factors and geographical features have differentially affected the genetic diversity across the regional frog assemblage. Much more complex models (i.e., considering multiple drivers), beyond simple vicariance of Caribbean and Pacific lineages, are needed to better understand the evolutionary history of ICA’s diverse biotas.
Similar content being viewed by others


Introduction
Isthmian Central America (ICA), including Costa Rica and Panama, is one of the most diverse regions in the globe (Myers et al. 2000). Inserted into the Mesoamerican hotspot of biodiversity, this region roughly covers 0.1% of Earth’s land surface, yet harbors an immense number of species. For Costa Rica alone, estimates for some taxonomic groups reach 4–10% of the global biodiversity, and Panama could be even more diverse (reviewed by Bagley and Johnson 2014). Some examples of highly diverse clades in the region include birds (>1000 spp), amphibians (>300 spp), nonavian reptiles (~500 spp), insects (>300,000 spp), and vascular plants (>20,000 spp) (Anger and Dean 2010; Garrigues and Dean 2014; Frost 2019).
ICA has a relatively recent geological origin, although the precise timing of the formation of the region’s major landscape features is debated (Montes et al. 2012; O’Dea et al. 2016). Historically, the most accepted hypothesis was that the Isthmus of Panama closed relatively recently, around 3–4 million years ago (Ma) (Coates et al. 1992). Evidence supporting this idea has been based most convincingly on divergence times of marine organisms separated by the Isthmus and several fossils found at the Panama Canal Zone (Keigwin 1978; O’Dea et al. 2016). However, the results from recent studies using petrographic, geochronological, and thermochronological data suggest that the Isthmus of Panama closed much earlier, around 10–15 Ma (Montes et al. 2012). An earlier date is also supported by some fossil evidence (Kirby et al. 2008; Bloch et al. 2016) and molecular phylogenetic studies showing early dispersal between North and South America around 10 Ma (Pinto-Sánchez et al. 2012; Bacon et al. 2015).
Independently of the debate over the precise date of the closure of the Isthmus of Panama, this geological formation facilitated one of the greatest biogeographic events of the Cenozoic, the Great American Biotic Interchange, a bidirectional dispersal of terrestrial mammals between the previously isolated North and South American landmasses (Marshall 1988; Webb 2006). Evidence of intercontinental dispersal in groups such as frogs, birds, and insects also highlights the important influence of the rise of the Isthmus in the assembly of the regional biota (Stehli and Webb 1985; Webb 2006; Weir et al. 2009; Pinto-Sánchez et al. 2012; Wilson et al. 2014) As a consequence, biodiversity in ICA is largely constituted by older northern plus southern lineages that arrived after the completion of the land bridge (Rich and Rich 1991; Savage 2002). Remarkably, the Isthmian fauna also has a strikingly high number of endemics (e.g., Kluge and Kessler 2006; Savage and Bolaños 2009; Bogarín et al. 2013; Garrigues and Dean 2014), including old lineages (Wang et al. 2008a). This high endemism highlights the importance of in situ diversification contributing to the conformation of the regional biota.
The complex tectonic and geological history of ICA resulted in a steep topography that encompasses a wide variety of habitats and climatic regimes over a small surface (Weyl 1980; Gabb et al. 2007). For example, in a span of less than 500 km along the Pacific coast of Costa Rica, the mean annual precipitation varies from ~1800 mm in some areas of the northern dry forest of Guanacaste to nearly 5000 mm in the very humid rainforests of the Osa Peninsula (Holdridge 1987; Bolaños et al. 2005). The physiography of Costa Rica in particular is characterized by NW-trending volcanic cordilleras that can reach altitudes over 3000 m in several peaks (Fig. 1) (Luteyn 1999). Such mountain systems are bisected by valleys that result in a mosaic of sky islands along with steep altitudinal gradients, microclimates, and diverse vegetation zones (Marshall 2007; Bagley and Johnson 2014). In addition, climatic and sea-level fluctuations during the Quaternary (Horn 1990; Islebe et al. 1996) likely promoted species-range contractions and expansions throughout this complex landscape, imposing or removing barriers to gene flow, and potentially playing a central role in the diversification processes (Ramírez-Barahona and Eguiarte 2013; Paz et al. 2019). The climatic dynamics associated with glacial cycles during the Pleistocene are thought to be responsible for shaping the distributional patterns of montane assemblages in the region (Savage 2002; Streicher et al. 2009). Given this historical context, allopatric speciation through vicariant events promoted by physical barriers (Mayr 1942) or parapatric speciation across environmental gradients (Doebeli and Dieckmann 2003) may have occurred extensively in the region, although strong evidence of the latter is still scarce.
Considering its recent geologic history and high levels of richness and endemism, ICA represents an ideal natural laboratory to study the role that landscape features have played in driving the early phases of genetic differentiation. One way to understand how such factors have influenced in situ diversification in ICA is to study the relationship between geographic or environmental variables and intraspecific genetic divergence. The discipline of landscape genetics combines advances in molecular tools and spatial statistical analyses to assess how the landscape structures populations (Manel et al. 2003; Manel and Holderegger 2013). In the simplest of the models studied under this approach, isolation by distance (IBD), a homogeneous landscape is assumed, and genetic differentiation is expected to increase monotonically with geographic distance due to limits in the per-generation dispersal distance (Wright 1943; Slatkin 1993).
In heterogeneous landscapes, however, dispersal may be more restricted due to the effect of barriers imposed by complex topographies and their associated climatic gradients (Manel and Holderegger 2013). Consequently, genetic isolation is expected to be more closely associated with landscape heterogeneity than geographical distances alone. A model of isolation by resistance (IBR) accounts for the reduced dispersal among populations caused by the relative unsuitability or “friction” presented by heterogeneous environments that may intervene between two populations (McRae 2006). Alternatively, the isolation-by-environment (IBE) model has been recently proposed to describe a process in which genetic isolation between two populations increases with local environmental differences, independently of the resistance or distance imposed by the landscape found between them (Wang and Bradburd 2014).
Quantifying the effects of IBD, IBR, and IBE in promoting genetic divergence within species can aid our understanding of the forces driving genetic diversity, species richness, and endemism in the ICA biota. Moreover, comparing the relative roles of these potential drivers of contemporary genetic variation across multiple codistributed species will reveal common spatial or historical causes behind potentially shared patterns (Feldman and Spicer 2006). As demonstrated in many previous studies, amphibians provide a good model system in comparative phylogeography because they tend to show a marked imprint of the effects of historical, ecological, and geographic factors in driving genetic divergence among populations (Beebee 2005; Smith and Green 2005; Wollenberg et al. 2011; Cruz-Piedrahita et al. 2018).
We hypothesize that spatial genetic variation within co-occurring amphibian species will reflect and reveal similar historical, ecological, and geographic drivers. We predict that species with overlapping distributions will display congruent responses to shared landscape-scale drivers of genetic variation (Avise 1992; Riddle et al. 2000; Feldman and Spicer 2006). Alternatively, variation among species in ecology and life history can result in independent patterns of genetic structuring in codistributed species (Crawford et al. 2007). In this case, environmental factors will differentially impinge upon species dispersal patterns, creating varied amounts of genetic structure in different species, depending on their habitat requirements (Michaux et al. 2005; Steele and Storfer 2007; Paz et al. 2015). For example, larger species could be less affected by topographic barriers due to their dispersal capabilities, while those species with wider geographic ranges and physiological tolerances could be less affected by climatic variation across the region.
To test this, we compared how landscape features of ICA predict patterns of variation in the 16S fragment within nine amphibian species with overlapping geographical distributions. To this end, we quantified the relative role of isolation by geographic distance (IBD), isolation by resistance due to topography (IBRtopo), isolation by resistance due to habitat suitability (IBRsuit), and isolation by environment (IBE) (e.g., local adaptation) in shaping genetic divergence within each species and tested the following predictions:
-
(1)
Distance alone (IBD) determines conspecific genetic variation. Linear geographic distance predicts genetic divergence among individuals.
-
(2)
Prominent mountains are major vicariant barriers (IBRtopo). Dispersal limitation imposed by topographic complexity predicts within-species genetic structure.
-
(3)
Unsuitable climatic conditions represent dispersal barriers among sampling localities (IBRsuit). Isolation caused by the patchy distribution of suitable habitats in the complex landscapes of the region shapes patterns of conspecific genetic divergence.
-
(4)
Habitat heterogeneity promotes local adaptation within distinct environments, and thus genetic isolation and divergence among conspecific populations (IBE). Differences among sampled sites in local climatic conditions predict patterns of genetic divergence.
-
(5)
Codistributed species will show similar relationships between genetic and geographic distance or spatial environmental variation. In contrast, idiosyncratic responses are predicted only to the extent that interspecific differences in life-history traits mediate the interaction between the environment and the organism’s dispersal potential.
Materials and methods
Study species
A portion of the DNA sequences used in this study represent new data derived from tissues previously deposited in the Herpetology Collection of the Museo de Zoología, Universidad de Costa Rica (hereafter UCR). With these samples, we generated an original dataset of mitochondrial DNA (mtDNA) sequences from 350 specimens, corresponding to 95 nominal species sampled from localities across Costa Rica as part of a barcoding survey of Costa Rican amphibians. From this dataset, we discarded potential cryptic species, and selected nine amphibian species with ample geographic sampling and that showed contrasting life histories, including seven anurans and two salamanders, represented by 37 individuals. These species belong to the families Plethodonthidae (two species of lungless salamanders), Hylidae (three species of tree frogs), Centrolenidae (two species of glass frogs), Craugastoridae (one species of rain frog), and Bufonidae (one toad species). These species have a variety of habits, reproductive strategies, body sizes, dispersal capabilities, and distribution ranges (Table 1). For example, cane toads (Bufonidae) are bigger, and have large clutch sizes. The two plethodontid salamanders have narrow distributions limited to the Pacific versant of the study region, and are poor dispersers. The direct-developing craugastorid frog species is less dependent on water availability, while centrolenid glass frogs are riparian species, and the terrestrial habitats among the watersheds may act as barriers. These features provide us with a variety of scenarios to test how species with different life histories respond to the same drivers of genetic differentiation.
We supplemented these newly generated data with GenBank data from orthologous DNA sequences for our nine study species, totaling 126 individuals sampled from Costa Rica and Panama, where we focused the questions of this study. The final dataset combining the newly generated sequences with the public data contained DNA data on a fragment of the 16S ribosomal RNA gene from 163 individuals. In Table S1 we provide species names, GenBank accession numbers, localities, and coordinates for each DNA sequence used in the study. The alignments for these sequences are available at Dryad repository.
While genome-wide markers would be preferable, mtDNA is the single-best standardized marker in multispecies comparative studies that focus on low-level divergence (not polymorphism) where the rapid substitution rate of mtDNA presents a distinct advantage in inferring genealogies, while the conservative structure of the mitochondrial genome provides standardized markers across diverse species (Carnaval et al. 2009; Wang 2013; Paz et al. 2015). Combining these features with the wide availability of public 16S sequence data, our novel nine-species comparative dataset presents a valuable opportunity for testing our hypotheses on the divers of genetic divergence in tropical tetrapods. While the genealogical information provided by any single locus may be unrepresentative of the genome as a whole, mtDNA in particular has some advantages of single nDNA markers. Being haploid and maternally inherited lowers the probability of incomplete lineage sorting by at least fourfold, if not more (Hudson and Coyne 2002). Also, while gene trees tend to overestimate population divergence times under a model of no migration, mtDNA does so less than nDNA due to the larger effective population size of the former markers (Edwards and Beerli 2000).
Sequencing methods
For the newly generated data, we carried out DNA extraction on a BioSprint 96 (Qiagen) robotic extractor based on magnetic beads, including digestion with proteinase K (0.4 mg/mL) at 55 °C. We amplified the slowly evolving 16S mitochondrial gene, following Crawford et al. (2010). This gene fragment was amplified and sequenced with primers 16SB-H (aka, 16Sbr-H) (5′-CCG GTC TGA ACT CAG ATC ACG T-3′) and 16SA-L (aka, 16Sar-L) (5′-CGC CTG TTT ATC AAA AAC AT-3′) (Kessing et al. 2004). PCR products were cleaned using ExoI and SAP enzymes (Werle et al. 1994) with Sanger sequencing products run on ABI 3130 automated sequencers. We performed all enzymatic and sequencing reactions in a high-throughput 96-well format. To confirm base calls, we sequenced the gene bidirectionally. The sequences were aligned using default parameters in MUSCLE (Edgar 2004, https://www.ebi.ac.uk/Tools/msa/muscle/) and reviewed by eye.
Potential cryptic species
Preliminary genetic analyses showed that some museum voucher specimens were misidentified, a common mistake in mega-diverse regions such as ICA, where a high number of sister species maintain morphological stasis after speciation (Padial and De La Riva 2009; Funk et al. 2012). After morphological species identification, we confirmed identification of DNA sequences using the Automated Barcode Gap Discovery (ABGD) algorithm (http://www.abi.snv.jussieu.fr/public/abgd/), which uses the distribution of pairwise genetic distances between the aligned DNA sequences to identify clusters of DNA sequences that may correspond to named or potential species (Puillandre et al. 2012). This method statistically infers multiple potential barcode gaps or thresholds, and partitions the sequences such that the distance between two sequences taken from distinct clusters is larger than the barcode gap. The method performs well in terms of speed and success in species identification compared with other DNA barcode algorithms (Paz and Crawford 2012). We processed the 16S alignments in ABGD assuming a Kimura 2-parameter (K2P) nucleotide substitution model (Kimura 1980) and the following settings: prior for the maximum value of intraspecific divergence between 0.001 and 0.1, with ten recursive steps within the primary partitions defined by the first estimated gap, and a gap width of 1.0 K2P is the standard model of DNA substitution for barcode studies performed, as well as other more complex models in identifying specimens (Collins and Cruickshank 2012). Even though there are not many DNA substitution models available in ABGD, species identification success rate is unlikely to be affected by the choice of substitution model (Collins et al. 2012).
To minimize potential taxonomic misidentifications within our nominal species, we confirmed that divergent, conspecific DNA sequences in sympatry showed K2P genetic divergence below 2.5% for 16S. This threshold was conservative compared with thresholds proposed by Vences et al. (2005) and Fouquet et al. (2007).
Estimation of genetic distances and gene trees
We estimated genetic distances in MEGA 4.0 (Tamura et al. 2013) invoking the pairwise-deletion option, thus including sites with gaps when the two individuals being compared had nucleotide data for that site. For each species, we calculated corrected genetic distances assuming the model of evolution determined as the best-fit model of nucleotide substitution for each set of conspecific sequences using MEGA (Tamura et al. 2013). Because we used a landscape genetics approach where the individual sequence is the unit of analysis (Balkenhol et al. 2008), we did not need to identify populations as required for other approaches to estimating genetic divergence, such as FST (Wright 1951). Details on the sizes of the DNA fragments utilized mean genetic distances, and substitution models for each species are provided in Table 2.
Gene trees were generated separately for each species using the maximum likelihood (ML) method implemented in the RAxML program, version 8.0 (Stamatakis 2014). We used the same substitution models mentioned above. Bootstrap values were calculated with 1000 pseudoreplicates. To delimit species, we used the ML trees as input and applied a Poisson tree process (PTP) model. This method identifies significant changes in the rate of branching in a phylogenetic tree using the number of substitutions, which are expected to be higher between species than within species (Zhang et al. 2013). PTP is a phylogeny-aware method that provides an objective estimation of phylogenetic entities based on branch-length dynamics instead of grouping just considering sequence similarities (Luo et al. 2018). Following a conservative approach, we report as genetic groups those clades with node values greater than 0.01, and ran an analysis of each species using the Bayesian implementation of the PTP model available in the webserver (http://species.h-its.org/ptp/) for 100,000 generations, with thinning = 100 and burn-in = 10%.
Estimation of geographic, resistance, and environmental distances
To compare the relative importance of different landscape variables in explaining genetic divergence within each species, we estimated landscape-derived distances, including linear geographic distance (IBD), resistance distances that accounted for topography (IBRtopo) and climatic suitability (IBRsuit), as well as local environmental dissimilarity (IBE) between all pairwise combinations of localities within each species. To test for IBD, we estimated linear horizontal geographic distances as pairwise Euclidean distances (in km) between the geographic position of each pair of individuals of each species using the R package, raster (Hijmans and van Etten 2010). To test for IBR, we estimated friction layers (derived from topography and environmental suitability) based on a circuit-theory approach conducted in Circuitscape V.4.0 (Shah and McRae 2008). For topography, we created a layer that quantifies local terrain complexity. We used a raster grid of elevation, with a resolution of 30 arcsec (~0.8 km at the equator), to estimate the standard deviation of a set of 25 adjacent cells to obtain a value of topographic complexity for cells of ~5-km resolution (the same resolution used for the species distribution models (SDMs)). This measure describes well the role of mountains as dispersal barriers as it is highly correlated with maximum altitude on each grid cell. Consequently, higher resistance values are associated with major mountain formations (Fig. S1). For environmental suitability, we used the inverse of a suitability derived from an SDM for each species, assuming that areas with low suitability have higher resistance for dispersal (Wang et al. 2008a, 2008b).
To construct SDMs, we gathered additional occurrence data across the entire distribution of each species, from the Global Biodiversity Information Facility (https://www.gbif.org/) and geographic records from the UCR. We generated SDMs using bioclimatic variables available at worldclim.org (2.5 arcmin resolution ~5 km at the equator) for current conditions. We excluded variables after assessing multicollinearity with a Variance Inflation Factor (VIF) analysis using the R package usdm (Naimi et al. 2014). This analysis quantifies the overall correlation of each variable with all others (Stine 1995). Collinearity is present when the VIF for at least one independent variable is large; then, we iteratively remove collinearity among bioclimatic variables by calculating VIF of variables against each other. To this, we used a value of 10 as the cutoff, as used in previous studies (Zuur et al. 2010). We created all models with a general-purpose machine-learning algorithm (MaxEnt) implemented in the R package, dismo (Hijmans et al. 2012), after parameter tuning and evaluation conducted in the R package, ENMeval (Muscarella et al. 2014; see Supplementary material). The performance of different SDM algorithms is contentiously debated in the literature, and different authors use different approaches (Hernandez et al. 2006; Li and Wang 2013). Fortunately, the relative performance of different algorithms is not a central issue for our implementation of SDMs. Rather than building models to predict what variables may determine species limits, our goal is only to generate surfaces of relative suitability for each species to then estimate the degree of isolation between localities. As long as our approach is relatively robust to assumptions, our analysis is not significantly affected by algorithm performance. Moreover, recent evidence comparing ensembles and single-algorithm methods, demonstrates that MaxEnt produces predictions of similar accuracy than those produced by ensembles, but reduces computational time and simplifies interpretations (Kaky et al. 2020).
To test for IBE, we estimated environmental dissimilarity between pairs of sampling localities. For each locality, we extracted the values for the 19 bioclimatic variables available in the Worldclim dataset (worldclim.org) at 30-arcsec resolution (~1 km). In this case, we kept all variables to have a more detailed estimation of the local climates, regardless of collinearity. We estimated Euclidean pairwise distances using the function ‘dist’ in R, which computes distances between the rows of a multivariate matrix.

The relief is drawn over a digital elevation model derived from the NASA Shuttle Radar Topographic Mission (SRTM). The highest elevations in the region are situated in the Talamanca range and overpass 3000 m (Marshall 2007).
Quantifying the relative effects of IBD, IBR, and IBE
We tested for possible associations between genetic distances and each of the four types of distances that we quantified (IBD, IBRtopo, IBRsuit, and IBE), using Multiple Matrix Regression with Randomization Analysis (MMRR), which uses randomized permutation to account for the nonindependence among pairwise distances (Wang 2013). MMRR estimates the effect of each variable as beta coefficients, allowing simultaneous comparisons among them. Because all estimated distances are at different scales, we standardized them (mean = 0, variance = 1) to facilitate the interpretation of the results. We performed the MMRR analysis with 10,000 permutations using an R function provided by Wang (2013).
We implemented a Generalized Dissimilarity Modelling (GDM) analysis to further explore how our independent variables may relate to observed genetic differentiation in each species. This is a multivariate technique that allows modeling a single response as a function of distance matrices for any number of explanatory variables (Manly 1986). GDM approach can fit nonlinear relationships of environmental variables to biological variation using I-spline functions (Ferrier et al. 2007). The spline plots provide insights into the total magnitude of biological change (i.e., genetic divergence) as a function of each gradient, and where, along each gradient, those changes are most pronounced. For each species, we fit a GDM using genetic distance as the dependent variable, and as predictor variables the distances quantified with the different types of isolation (IBD, IBRtopo, IBRsuit, or IBE). We plotted the I splines to assess how magnitudes and rates of genetic differentiation varied along the gradients represented in our independent (predictor) variables. To select the best combination of predictor variables, we used a permutation-based backward elimination (as detailed in Ferrier et al. 2007). This procedure calculates the unique contribution of each predictor to the total explained deviance, and discards the variable that least contributes to fit a new GDM using a reduced set of n–1 predictors. We used 500 permutations and repeated the method until all variables retained in the final model made significant, unique contributions to the explained deviance (P ≤ 0.05). We performed these analyses with the R package, gdm (Fitzpatrick et al. 2020).
One potential limitation of the IBD, IBR, and IBE analyses is the use of predictor variables covering both historical and present-day processes. For example, while topographic variation and geographic distances mirror both historical and ongoing processes, habitat suitability estimations reflect contemporary climatic niches. For the latter case, we attenuate this issue by projecting our suitability models to the Last Glacial Maximum (LGM). However, the IBR matrices estimated for present and past time periods were correlated, suggesting relative stability of environments at the regional level (of the ICA) across the extremes of interglacial and glacial periods (today and the LGM). Therefore, we decided to use only the suitability predictions for current climate conditions. This will facilitate posterior interpretations. Additionally, we tested for collinearity among variables conducting Mantel tests (Table S2).
Results
Intraspecific patterns of genetic differentiation
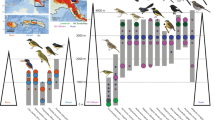
We found overall intraspecific genetic distances ranging between 0.36% in Rhinella horribilis and 4.02% in Pristimantis ridens (Table 2). For the two salamander species in our dataset, we found a major genetic break between populations of Bolitoglossa lignicolor in the central Pacific region and those extending from the southern Pacific of Costa Rica to Peninsula de Azuero in Panama (Fig. 2). In the case of Oedipina alleni, we found major differences between an individual from the Peninsula de Osa and the rest of sampled localities (Fig. 2).

Each panel presents the mtDNA gene tree for a given species with major clades, given different colors, mapped to sampling localities (dots) over a digital elevation map of ICA with elevations similar to Fig. 1, but with increasing elevations indicated by warmer colors (sea level: blue; highest peaks: red). Scale bars are gene tree distances given in units of substitutions/site. Panels (a, b) are salamander species and likely poor dispersers. Panel (c) is a direct-developing frog common in wet forests across the study area. Panels (d, e) are glass frogs mostly associated to riparian habitats. Panels (f–h) are tree frog species that reproduces in ponds and streams. Panel (i) is the cane toad, the larger species in the study and the one with the larger clutch size along with (g). Congruence among species’ phylogeographic structure is minimal.
For the direct-developing frog, P. ridens, we found support for a clade containing all Costa Rican samples, two divergent clades from the Coclé region, and a clade containing populations from eastern Panama. In the glass frogs (Centrolenidae), we found evidence for genetic divergence between populations of Sachatamia albomaculata in the Caribbean of Costa Rica and the Pacific of Costa Rica plus Panama (Fig. 2). In Espadarana prosoblepon, we found four clades: one from the Darien region of Panama, a second containing samples from localities near the Panama Canal, a third grouping ranging from El Copé in central Panama to the Pacific versant of Costa Rica (from Tilarán to the southern foothills of Talamanca), and the final clade containing an individual from the Central Valley of Costa Rica (Fig. 2).
Among the hylid species, Dendropsophus ebraccatus was represented by samples only from Costa Rica, which showed a major “north–south” genetic split between the southern Pacific plus central Caribbean versus central Pacific plus the northern portion of the Caribbean versant (Fig. 2). In Smilisca phaeota, samples grouped into two major clades mostly divided by the Central Cordillera and the Talamanca Cordillera; these clades overlap in distribution in the lower mountain passes between the Tilarán Cordillera and the Central Valley of Costa Rica (Fig. 2). In Agalychnis callidryas, we found that Costa Rican samples belong to three clades: one formed by only Pacific samples, a second including mostly Caribbean samples plus two samples from the northern Pacific of Costa Rica, and a third group containing samples from the Coclé region of Caribbean Panama and the southern Caribbean of Costa Rica (Fig. 2). Samples from central Panama formed a fourth clade. Finally, in the cane toad, R. horribilis, we detected two clades that do not show a clear geographic pattern.
Relative role of IBD, IBRtopo, IBRsuit, and IBE on genetic differentiation
From the MMRR analyses, we found that in five out of nine species, different factors tested here significantly explained the patterns of genetic differentiation, depending on the species (Table 3). None of the models tested explained patterns of genetic differentiation in the salamander species (O. alleni and B. lignicolor), the hylid frog S. phaeota, or the toad R. horribilis. In the species where the model significantly explained genetic divergence among populations, we found idiosyncratic responses to the predictors tested. Geographic distance (IBD) had a significant effect on the genetic differentiation in P. ridens (βIBD = 0.648, P < 0.001); topography (IBRtopo) explained most genetic divergence for A. callidryas (βIBRtopo = 0.580, P < 0.001) and S. albomaculata (βIBRtopo = 0.580, P < 0.001). Among the factors tested, isolation due to climatic suitability (IBRsuit) had the greatest effect on the genetic divergence for D. ebraccatus (βIBRsuit = 0.998, P < 0.001). Local environmental dissimilarity (IBE) was involved in explaining genetic variation within E. prosoblepon (βIBE = 0.486, P < 0.001).
From the GDM results, we confirmed that the response of each species to the factors considered is idiosyncratic, including the magnitude of the gradient of variation of each predictor. We illustrate this heterogeneity by presenting the spline plots for the two variables that better explained the patterns of genetic differentiation in the species where the model was significant (Fig. 3). Spline plots for all variables tested for each species are available in the Supplementary Material (Fig. S1). Details of the SDMs generated to estimate IBRsuit and the respective projections on the study areas are available in the Supplementary Material (Table S3 and Fig. S2).

The figure shows the results only for the five species that the factors tested significantly explained the patterns of genetic differentiation. The maximum height reached by each curve shows the total amount of genetic differentiation associated with the respective variable, holding all other variables constant. The shape of each function shows how the rate of genetic differentiation varies along the variation gradient of a given variable. The different combinations of colors show how each species responds differently to the variables tested. Photos by Víctor Acosta (P. ridens, E. prosoblepon, A. callidryas, and S. albomaculata) and Brian Gratwicke (D. ebraccatus).
Discussion
We quantified how different components of the landscape have restricted gene flow and promoted diversification across several amphibian species in ICA. In a majority of the species studied here, at least one of our measures of geographic distance or environmental heterogeneity (i.e., based on topography, habitat suitability, or climatic dissimilarity) significantly explained intraspecific patterns of genetic variation. However, instead of finding a single, major factor driving such patterns across taxa, our results suggest idiosyncratic responses among species, likely related to differences in their life histories. The novelty of our study lies in the generation of new sequences to compile a dataset comprising over 160 individuals of multiple codistributed species and its use in a comparative framework. This is especially relevant in ICA, a highly biodiverse region that has been poorly studied from this perspective (Bagley and Johnson 2014). We test the influence of each predictor of genetic variation (Wang 2013) and quantify the effect size of environmental variation in each predictor on genetic divergence (Ferrier et al. 2007). This was key in our attempt to assess whether genetic structure across all species is similarly influenced by key isolating factors acting in ICA, independently of their temporal reach.
Comparing our results with previous phylogeographic studies of amphibians of ICA, we note that these earlier studies also show few spatial commonalities among species (Weigt et al. 2005; Crawford et al. 2007; Wang et al. 2008a, 2008b; Robertson et al. 2009). Previous studies have identified at least 31 phylogeographic breaks from mtDNA markers in plants, insects, and vertebrates (Bagley and Johnson 2014) from across this ~131,000-km2 region. Population divergences within the regional frog assemblage have resulted from one or multiple historical events during the Pliocene–Pleistocene (Bagley et al. 2018). Our findings add a new perspective to our understanding of the region’s complex evolutionary history by showing that such events are likely driven by multiple forces acting on the ICA and having differential effects on genetic divergence within species. Below, we discuss these results recognizing the strength of the dataset compiled for multiple co-occurring species in a region still understudied in terms of comparative phylogeography, but also bearing in mind the limitations associated with our genetic sampling and the time-scale differences among variables tested.
Geographic distance as the predictor of genetic divergence
Although geographic distance (IBD) was not expected to be the best predictor of genetic divergence due to the region’s complex topography and environmental heterogeneity, this variable explained significantly the genetic structure within E. prosoblepon and P. ridens. Previous work in the region has also found phylogeographic breaks not necessarily associated with obvious physical or climatic barriers (Bagley and Johnson 2014). For example, the Bocas del Toro region is an important filter barrier on the Caribbean coast that separates northwestern from southeastern populations, despite the continuous lowland wet forest habitat connecting Costa Rica and Panama (Crawford et al. 2007; Robertson et al. 2009). This could explain how our samples of P. ridens from the Pacific and the Caribbean versants of Costa Rica showed even less divergence between each other than between them and Panamanian samples collected east of the Bocas del Toro region. In this species, Panamanian lineages show long-term geographic stasis in contrast to the rapid geographic expansion that likely occurred from Costa Rica to Honduras (Wang et al. 2008a, 2008b), supporting both the differentiation of Panamanian samples and the absence of structure in the Costa Rican portion of P. ridens samples.
Dispersal within streams and among adjacent headwaters can occur in riparian species, such as the here-studied glass frog E. prosoblepon (Robertson et al. 2008). Despite the individual’s strong site fidelity (Jacobson 2018), their capacity for dispersal maintains genetic homogeneity at the fine scale (2.5–5 km2), including among streams, while terrestrial gene flow naturally decreases with distance (Robertson et al. 2008). Our results support this idea and suggest that geographic distance between watersheds explains genetic differentiation better than topographic or climatic barriers within a specific watershed, which could be more permeable through riparian corridors for species associated with these habitats.
Major topographic barriers limiting gene flow
The mountainous relief of ICA caused by volcanic and tectonic orogeny (Gabb et al. 2007) may impose a major physical barrier (IBRtopo) to the dispersal and gene flow of many species. Intraspecific genetic divergence is expected to be greater in areas of higher topographic complexity (Guarnizo and Cannatella 2013). We found this pattern in the red-eyed tree frog, A. callidryas, a lowland species whose genetic divergence is more evident between populations isolated by major orogenic barriers, such as the Talamanca Cordillera, and decreases between northeastern and northwestern populations where topographic barriers are less continuous (e.g., the Tilarán and Guanacaste cordilleras) (Robertson et al. 2009). Similarly, the genetic divergence between individuals of S. albomaculata on either side of the central mountain ranges of Costa Rica reflects the importance of mountainous barriers in restricting gene flow for some lowland species. This glass frog is more common below 1000 m (Kubicki 2007), which limits potential dispersal across most mountain ranges in the region, especially across those with continuous elevations above 1500 m from Central Cordillera in Costa Rica to the Tabasará Range of western Panama (aka the eastern Talamanca Cordillera) (Janzen 1991). These results highlight the role of high-elevation Cordilleras as important dispersal barriers for lowland organisms.
In the region, various closely related species distributed on each side of major mountain systems may have experienced isolation promoted by the topography. Some examples include the frogs Craugastor stejnegerianus and C. persimilis or Phyllobates lugubris and P. vittatus (Savage 2002; Crawford 2003; Grant et al. 2006), and the bushmaster snakes Lachesis stenophrys and L. melanocephala (Alencar et al. 2016). These patterns occur even in groups with higher dispersal abilities, such as the birds Amazilia decorata and A. amabilis, Cotinga ridwayi, and C. amabilis, or Manacus candei and M. aurantiacus (Prum et al. 2000; Brumfield and Braun 2001; Stiles et al. 2017). Isolation between Pacific and Caribbean groups is more evident in eastern Costa Rica and western Panama, where topographic barriers become stronger as mountains reach >3500-m elevation in the Talamanca Cordillera (de Boer et al. 1995). In contrast, in Tilarán and Guanacaste cordilleras to the north, where elevations are lower and mountain passes wider (Janzen 1991), the relevance of topography on genetic differentiation might be minor and replaced by other factors such as geographic distance or climate.
Influence of habitat suitability on dispersal and genetic differentiation
Climatic barriers imposed by differential habitat suitability (IBRsuit) may shape genetic structure by limiting exchange of individuals and genes among populations. In ICA, such patterns highlight the importance of within-versant climatic variation representing barriers of unsuitable habitats to clades from the central and southern Pacific regions, for example (Holdridge 1987). We found that environmental resistance associated with less-suitable regions best explains genetic differentiation in D. ebraccatus (Fig. 3). In this species, our sampling is restricted to Costa Rica, but includes individuals from both versants. Interestingly, we found samples from both the Central Pacific and the Caribbean grouped in the same clade (Fig. 2) that was sister to a clade from the southern Pacific. Previous evidence shows the sensitivity of this same species to climatic barriers in the divergence of lineages associated with the western Panama Isthmus break (WPI, Bagley et al. 2018), a zone characterized by predominantly semiarid conditions since 12–14 Ma (Crawford et al. 2007; Wang et al. 2008a, 2008b). The barrier detected here seems to be associated with the Central Pacific transition between dry and humid conditions, which represents a well-defined boundary for biogeographical provinces of ichthyofauna and herpetofauna (Bagley and Johnson 2014). The region’s climatic heterogeneity thus also has an important influence as a driver of genetic differentiation for some representatives of the biota of ICA.
The role of local adaptation in patterns of genetic divergence
Adaptation to local climatic conditions along altitudinal or latitudinal gradients could also promote genetic differentiation in this region. We evaluated climatic heterogeneity (IBE), measuring how different two localities are, independent of the intervening landscape (Wang and Bradburd 2014). Our data show that this factor has a contribution in explaining genetic structure in E. prosoblepon. For this species, we detected divergence over short distances, for example, between the Central Valley, the Tilarán Cordillera, and the Central Pacific of Costa Rica.
One of the most striking characteristics of ICA is the variety of climates that converge in such a small area (Coen 1991). For example, transitions between semiarid and very humid conditions occur in a couple of 100 km in the Pacific of Costa Rica, while the temperature may change significantly over shorter distances along elevation gradients in the mountain systems (Coen 1991; Savage 2002). Variation in species-specific tolerances and local adaptation to such diverse climates influence the distribution of several taxa in the region. Some examples occur even in groups considered good dispersers, such as birds (Garrigues and Dean 2014) and volant mammals like the vespertilionid bats Rhogeessa bickhami and R. io (Baird et al. 2012). Our analysis revealed a slight contribution of environmental distance (independent of geographic distance) in explaining the genetic structure in this species. Even slight changes in climatic regimes could drive local adaptation and isolation with evolutionary implications such as genetic differentiation for other species in the region.
Additional factors driving phylogeographic structure
Variation in phylogeographic structure among species of anurans can be explained in part by variation in life-history traits (Paz et al. 2015). As shown above, species respond idiosyncratically to each factor tested here, and for a few species, no factor was related to spatial patterns of genetic differentiation. Our full model containing all predictors and their interactions did not significantly explain the genetic divergence in the salamanders, B. lignicolor or O. alleni, in the masked tree frog, S. phaeota, or in the cane toad, R. horribilis. Perhaps not coincidently, for these species, we had lower sample sizes and, consequently, less predictive power (Table S2). Both salamander species are restricted to the Pacific slope of the study region, below 900 m (Savage 2002), which limits the opportunity to test for prominent topographic or climatic barriers along broad altitudinal gradients. However, these species also show several particularities that may account for such a lack of a significant relationship between our predictors and genetic structure.
Although our sampling of S. phaeota and R. horribilis covered a wide range of climatic conditions and elevations, we did not find evidence of a relationship between the factors tested here and spatial genetic variation in these two frog species. Both species have a large body size that may facilitate high dispersal ability, and they have wide distributions that reflect their broader intrinsic physiological tolerances, even in altered habitats (Savage 2002). These features, along with their high reproductive rate with thousands of eggs per clutch, may explain their distribution and persistence under heterogeneous environmental pressures (Hilje and Arévalo-Huezo 2012; McCann et al. 2014). Thus, barriers such as topography or environmental variation that affect other amphibian species may be more permeable for S. phaeota and R. horribilis. Our results support the hypothesis that variation among species in life-history traits drives the variable responses among species to a common landscape, although we must temper this conclusion by the fact that our genetic data came from a single marker. Still, intrinsic factors, such as body size and reproductive mode, may be as important as landscape features in driving lineage diversification (Wollenberg et al. 2011; Paz et al. 2015; Wollenberg Valero 2015).
In conclusion, interspecific differences in life-history traits could be responsible to mediate the interaction between the environment and the organism’s dispersal potential. Moreover, the convergence of abiotic factors that may act as drivers of isolation (i.e., complex topography and heterogeneous climates) and the co-occurrence of a heterogeneous biota with disparate life traits may have fueled the increased biodiversity across ICA. Different types of isolation, such as IBD and IBE (reviewed in Sexton et al. 2014) or IBR associated with topographic or climatic barriers (Oliveira et al. 2017a, 2017b), are known to promote genetic differentiation in different organisms. Our study particularly suggests that these forces may act simultaneously even in a relatively small region and differentially affect codistributed species. Certainly, species-specific life-history traits play a key role in how species respond to different drivers of isolation, and such interaction between organisms and their environment must also be considered when trying to understand patterns of genetic divergence (Paz et al. 2015; Rodríguez et al. 2015; Wollenberg Valero 2015). Although based on a single genetic marker, our findings suggest that complex processes, including several abiotic and geographical features, are necessary to explain in situ diversification and understand the evolutionary dynamics of the ICA’s amphibian community. We hope that this work motivates the development of further studies, including additional taxa and stronger molecular sampling to continue improving our understanding of the evolution of this region’s diverse biota.
Data availability
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.2jm63xsn0.
References
Alencar LRV, Quental TB, Grazziotin FG, Alfaro ML, Martins M, Venzon M et al. (2016) Diversification in vipers: phylogenetic relationships, time of divergence and shifts in speciation rates. Mol Phylogenet Evol 105:50–62
Anger GR, Dean R (2010) The birds of Panama: a field guide. Zona Tropical Publication, San José, Costa Rica
Avise JC (1992) Molecular population structure and the biogeographic history of a regional fauna: a case history with lessons for conservation biology. Oikos 63:62–76
Bacon CD, Silvestro D, Jaramillo C, Smith BT, Chakrabarty P, Antonelli A (2015) Biological evidence supports an early and complex emergence of the Isthmus of Panama. Proc Natl Acad Sci 112:6110–6115
Bagley JC, Hickerson MJ, Johnson JB (2018) Testing hypotheses of diversification in Panamanian frogs and freshwater fishes using hierarchical approximate Bayesian computation with model averaging. Diversity 10:1–25
Bagley JC, Johnson JB (2014) Phylogeography and biogeography of the lower Central American Neotropics: diversification between two continents and between two seas. Biol Rev 89:767–790
Baird AB, Marchán-Rivadeneira MR, Pérez SG, Baker RJ (2012) Morphological analysis and description of two new species of Rhogeessa (Chiroptera: Vespertilionidae) from the neotropics. Occas Pap Mus Tex Tech Univ 307:1–25
Balkenhol N, Cushman SA, Storfer AT, Waits LP (2008) Landscape Genetics. Concepts, Methods, Applications. Wiley-Blackwell, West Sussex, UK
Beebee TJC (2005) Conservation genetics of amphibians. Heredity 95:423–7
Bloch JI, Woodruff ED, Wood AR, Rincon AF, Harrington AR, Morgan GS et al. (2016) First North American fossil monkey and early Miocene tropical biotic interchange. Nature 533:243–246
de Boer JZ, Drummond MS, Bordelon MJ, Defant MJ, Bellon H, Maury RC (1995) Cenozoic magmatic phases of the Costa Rican island arc (Cordillera de Talamanca). Geol Soc Am Spec Pap 295:35–56
Bogarín D, Pupulin F, Arrocha C, Warner J (2013) Orchids without borders: studying the hotspot of Costa Rica and Panama. Lankesteriana 13:13–26
Bolaños R, Watson V, Tosi J (2005) Mapa ecológico de Costa Rica (Zonas de Vida), según el sistema de clasificación de zonas de vida del mundo de LR Holdridge), Escala 1:750 000. Centro Científico Tropical, San José, Costa Rica. Centro Científico Tropical, San José, Costa Rica
Brumfield RT, Braun MJ (2001) Phylogenetic relationships in bearded manakins (Pipridae: Manacus) indicate that male plumage color is a misleading taxonomic marker. Condor 103:248–258
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C (2009) Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science 323:785–789
Coates AG, Jackson JBC, Collins LS, Cronin TM, Dowtsett HJ, Bybell LM et al. (1992) Closure of the Isthmus of Panama: the near-shore marine record of Costa Rica and western Panama. Geol Soc Am Bull 104:814–828
Coen E (1991) Climate. In: Janzen DH (ed) Historia Natural de Costa Rica. Editorial de la Universidad de Costa Rica, San José, Costa Rica, p 822
Collins RA, Boykin LM, Cruickshank RH, Armstrong KF (2012) Barcoding’s next top model: an evaluation of nucleotide substitution models for specimen identification. Methods Ecol Evol 3:457–465
Collins RA, Cruickshank RH (2012) The seven deadly sins of DNA barcoding. Mol Ecol Resour 13:969–975
Crawford AJ (2003) Huge populations and old species of Costa Rican and Panamanian dirt frogs inferred from mitochondrial and nuclear gene sequences. Mol Ecol 12:2525–2540
Crawford AJ, Bermingham E, Carolina PS (2007) The role of tropical dry forest as a long-term barrier to dispersal: a comparative phylogeographical analysis of dry forest tolerant and intolerant frogs. Mol Ecol 16:4789–807
Crawford AJ, Lips KR, Bermingham E (2010) Epidemic disease decimates amphibian abundance, species diversity, and evolutionary history in the highlands of central Panama. Proc Natl Acad Sci 107:13777–13782
Cruz-Piedrahita C, Navas CA, Crawford AJ (2018) Life on the edge: a comparative study of ecophysiological adaptations of frogs to Tropical semiarid environments. Physiol Biochem Zool 91:740–756
Doebeli M, Dieckmann U (2003) Speciation along environmental gradients. Nature 421:259–264
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Edwards S, Beerli P (2000) Perspective: gene divergence, population divergence, and the variance in coalescence time in phylogeographic studies. Evolution 54:1839–1854
Feldman CR, Spicer GS (2006) Comparative phylogeography of woodland reptiles in California: repeated patterns of cladogenesis and population expansion. Mol Ecol 15:2201–2222
Ferrier S, Manion G, Elith J, Richardson K (2007) Using generalized dissimilarity modelling to analyse and predict patterns of beta diversity in regional biodiversity assessment. Divers Distrib 13:252–264
Fitzpatrick MC, Mokany K, Manion G, Lisk M, Ferrier S, Nieto-Lugilde D (2020) gdm: Generalized dissimilarity modeling. https://CRANR-project.org/package=gdm.
Fouquet A, Vences M, Salducci M-D, Meyer A, Marty C, Blanc M et al. (2007) Revealing cryptic diversity using molecular phylogenetics and phylogeography in frogs of the Scinax ruber and Rhinella margaritifera species groups. Mol Phylogenet Evol 43:567–582
Frost DR (2019) Amphibian species of the world: an online reference. Version 6.0 (Date of access). Electronic database accessible at. Amphib Species World an Online Ref Version 60.
Funk WC, Caminer M, Ron SR (2012) High levels of cryptic species diversity uncovered in Amazonian frogs. Proc R Soc B 279:1806–1814
Gabb WM, Lücke OH, Gutiérrez V, Soto G (2007) On the geology of the Republic of Costa Rica. (Transcription of the original manuscript by: Oscar H. Lücke, Viviana Gutiérrez & Gerardo Soto). Rev Geológica América Cent 203:103–118
Garrigues R, Dean R (2014) The Birds of Costa Rica. Zona Tropical Publication, San José, Costa Rica
Grant T, Frost DR, Caldwell JP, Gagliardo R, Haddad CFB, Kok PJR et al. (2006) Phylogenetic systematics of dart-poison frogs and their relatives (Amphibia: Athesphatanura: Dendrobatidae). Bull Am Mus Nat Hist 299:1–262
Guarnizo CE, Cannatella DC (2013) Genetic divergence within frog species is greater in topographically more complex regions. J Zool Syst Evol Res 51:333–340
Hernandez PA, Graham CH, Master LL, Albert DL (2006) The effect of sample size and species characteristics on performance of different species distribution modeling methods. Ecography 29:773–785
Hijmans RJ, van Etten J (2010) raster: Geographic analysis and modeling with raster data. R Packag version 1.
Hijmans RJ, Phillips S, Leathwick J, Elith J (2012) Package ‘dismo’. Species distribution modeling. R Packag version 08-11.
Hilje B, Arévalo-Huezo E (2012) Aestivation in the cane toad Rhinella marina Linnaeus 1758 (Anura, Bufonidae) during the peak of a dry season in a tropical dry forest, Costa Rica. Herpetol Notes 5:533–534
Holdridge LR (1987) Ecología basada en zonas de vida. Instituto Interamericano de Cooperación para la Agricultura (IICA), San Jose, Costa Rica
Horn S (1990) Timing of deglaciation in the Cordillera de Talamanca. Costa Rica Clim Res 1:81–83
Hudson RR, Coyne JA (2002) Mathematical consequences of the genealogical species concept. Evolution 56:1557–1565
Islebe GA, Hooghiemstra H, van’t Veer R (1996) Holocene vegetation and water level history in two bogs of the Cordillera de Talamanca, Costa Rica. Vegetatio 124:155–171
Jacobson S (2018) Reproductive behavior and male mating success in two species of glass frogs. Herpetologica 41:396–404
Janzen DH (1991) Historia Natural de Costa Rica. Primera (D Janzen, Ed.). Editorial San José, Universidad de Costa Rica, San José, Costa Rica
Kaky E, Nolan V, Alatawi A, Gilbert F (2020) A comparison between Ensemble and MaxEnt species distribution modelling approaches for conservation: a case study with Egyptian medicinal plants. Ecol Inf 60:101150
Keigwin LD (1978) Pliocene closing of the Isthmus of Panama, based on biostratigraphic evidence from nearby Pacific Ocean and Caribbean Sea cores. Geology 6:630–634
Kessing B, Croom H, Martin A, McIntosh C, Owen MW, Palumbi SP (2004) The Simple Fool’s Guide to PCR, Version 1. Department of Zoology, University of Hawaii, Honolulu, USA
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kirby MX, Jones DS, MacFadden BJ (2008) Lower Miocene stratigraphy along the Panama Canal and its bearing on the Central American Peninsula. PLoS ONE 3:e2971
Kluge J, Kessler M (2006) Fern endemism and its correlates: contribution from an elevational transect in Costa Rica. Divers Distrib 12:535–545
Kubicki B (2007) Ranas de Vidrio de Costa Rica, 1st edn. Instituto Nacional de Biodiversidad, Santo Domingo de Heredia, Costa Rica
Li X, Wang Y (2013) Applying various algorithms for species distribution modelling. Integr Zool 8:124–135
Luo A, Ling C, Ho SYW, Zhu CD (2018) Comparison of methods for molecular species delimitation across a range of speciation scenarios. Syst Biol 67:830–846
Luteyn J (1999) Paramos: a checklist of plant diversity, geographical distribution, and botanical literature. Mem N. Y Bot Gard 84:138–141
Manel S, Holderegger R (2013) Ten years of landscape genetics. Trends Ecol Evol 28:614–621
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197
Manly B (1986) Randomization and regression methods for testing for associations with geographical, environmental and biological distances between populations. Res Popul Ecol 28:201–218
Marshall JS (2007) The geomorphology and physiographic provinces of Central America. In: Bundschuh & Alvarado (eds) Central America: Geology, Resources and Hazards, vol 1, CRC Press, Florida, USA, pp 1–51
Marshall LG (1988) Land mammals and the Great American interchange. Am Sci 76:380–388
Mayr E (1942) Systematics and the origin of species, 1st edn. Columbia University Press, New York, NY
McCann S, Greenlees MJ, Newell D, Shine R (2014) Rapid acclimation to cold allows the cane toad to invade montane areas within its Australian range. Funct Ecol 28:1166–1174
McRae BH (2006) Isolation by resistance. Evolution 60:1551–1561
Michaux JR, Libois R, Filippucci MG (2005) So close and so different: Comparative phylogeography of two small mammal species, the Yellow-necked fieldmouse (Apodemus flavicollis) and the Woodmouse (Apodemus sylvaticus) in the Western Palearctic region. Heredity 94:52–63
Montes C, Cardona A, McFadden R, Morón SE, Silva CA, Restrepo-Moreno S et al. (2012) Evidence for middle Eocene and younger land emergence in central Panama: implications for Isthmus closure. Bull Geol Soc Am 124:780–799
Muscarella R, Galante PJ, Soley-Guardia M, Boria RA, Kass JM, Uriarte M et al.(2014) ENMeval: an R package for conducting spatially independent evaluations and estimating optimal model complexity for MaxEnt ecological niche models Methods Ecol Evol 5:1198–1205
Myers N, Mittermeier RA, Mittermeier CG, Fonseca GAB, da, Kent E (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858
Naimi B, Hamm NAS, Groen TA, Skidmore AK, Toxopeus AG (2014) Where is positional uncertainty a problem for species distribution modelling? Ecography 37:191–203
O’Dea A, Lessios HA, Coates AG, Eytan RI, Restrepo-Moreno SA, Cione AL et al. (2016) Formation of the Isthmus of Panama. Sci Adv 2:1–12
Oliveira BF, São-Pedro VA, Santos-Barrera G, Penone C, Costa GC (2017b) AmphiBIO, a global database for amphibian ecological traits. Sci Data 4:170123
Oliveira E, Martinez P, Sao-Pedro V, Gehara M, Burbrink FT, Mesquita D et al. (2017a) Climatic suitability, isolation by distance and river resistance explain genetic variation in a Brazilian whiptail lizard. Heredity 120:251–265
Padial JM, De La Riva I (2009) Integrative taxonomy reveals cryptic Amazonian species of Pristimantis (Anura: Strabomantidae). Zool J Linn Soc 155:97–122
Paz A, Crawford AJ (2012) Molecular-based rapid inventories of sympatric diversity: a comparison of DNA barcode clustering methods applied to geography-based vs clade-based sampling of amphibians. J Biosci 37:887–896
Paz A, González A, Crawford AJ (2019) Testing effects of Pleistocene climate change on the altitudinal and horizontal distributions of frogs from the Colombian Andes: A species distribution modeling approach. Front Biogeogr 11:e37055
Paz A, Ibáñez R, Lips KR, Crawford AJ (2015) Testing the role of ecology and life history in structuring genetic variation across a landscape: a trait-based phylogeographic approach. Mol Ecol 24:3723–3737
Pinto-Sánchez NR, Ibañez R, Madriñán S, Sanjur OI, Bermingham E, Crawford AJ (2012) The Great American biotic interchange in frogs: multiple and early colonization of Central America by the South American genus Pristimantis (Anura: Craugastoridae). Mol Phylogenet Evol 62:954–972
Prum R, Rice NH, Mobley J, Dimmick W (2000) A preliminary phylogenetic hypothesis for the cotingas (Cotingidae) based on mitochondrial DNA. Auk 117:236–241
Puillandre N, Lambert A, Brouillet S, Achaz G (2012) ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol 21:1864–1877
Ramírez-Barahona S, Eguiarte LE (2013) The role of glacial cycles in promoting genetic diversity in the Neotropics: The case of cloud forests during the Last Glacial Maximum. Ecol Evol 3:725–738
Rich PV, Rich T (1991) La ruta de dispersión centroamericana: Historia biotica y paleográfica. In: Janzen DH (ed) Historia Natural de Costa Rica. Editorial San José, Universidad de Costa Rica, San José, Costa Rica, p 13–34
Riddle BR, Hafner DJ, Alexander LF, Jaeger JR (2000) Cryptic vicariance in the historical assembly of a Baja California Peninsular Desert biota. Proc Natl Acad Sci USA 97:14438–14443
Robertson JM, Duryea MC, Zamudio KR (2009) Discordant patterns of evolutionary differentiation in two Neotropical treefrogs. Mol Ecol 18:1375–1395
Robertson JM, Lips KR, Heist EJ (2008) Fine scale gene flow and individual movements among subpopulations of Centrolene prosoblepon (Anura: Centrolenidae). Rev Biol Trop 56:13–26
Rodríguez A, Börner M, Pabijan M, Gehara M, Haddad CFB, Vences M (2015) Genetic divergence in tropical anurans: deeper phylogeographic structure in forest specialists and in topographically complex regions. Evol Ecol 29:765–785
Savage J, Bolaños F (2009) A checklist of the amphibians and reptiles of Costa Rica: additions and nomenclatural revisions. Zootaxa 2005:1–23
Savage JM (2002) The Amphibians and Reptiles of Costa Rica: a herpetofauna between two continents, between two seas. University of Chicago press, Chicago, Illinois, USA
Sexton JP, Hangartner SB, Hoffmann AA (2014) Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution 68:1–15
Shah VB, McRae BH (2008) Circuitscape: a tool for landscape ecology. Proc 7th Python Sci Conf. 7:62–65
Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47:264–279
Smith AM, Green DM (2005) Dispersal and the metapopulation paradigm in amphibian ecology and conservation: are all amphibian populations metapopulations? Ecography 28:110–128
Stamatakis A (2014) RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Steele CA, Storfer A (2007) Phylogeographic incongruence of codistributed amphibian species based on small differences in geographic distribution. Mol Phylogenet Evol 43:468–479
Stehli FG, Webb SD (eds) (1985) The Great American Biotic Interchange. Plenum Press, New York, NY (FG Stehli and SD Webb, Eds.)
Stiles G, Remsen JV, McGuire JA (2017) The generic classification of the Trochilini (Aves: Trochilidae): reconciling taxonomy with phylogeny. Zootaxa 4353:401–424
Stine R (1995) Graphical interpretation of variance inflation factors. Am Stat 49:53–56
Streicher JW, Crawford AJ, Edwards CW (2009) Multilocus molecular phylogenetic analysis of the montane Craugastor podiciferus species complex (Anura: Craugastoridae) in Isthmian Central America. Mol Phylogenet Evol 53:620–630
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Vences M, Thomas M, Van Der Meijden A, Chiari Y, Vieites DR (2005) Comparative performance of the 16S rRNA gene in DNA barcoding of amphibians. Front Zool 2:1–12
Wang I, Crawford AJ, Bermingham E (2008a) Phylogeography of the Pygmy Rain Frog (Pristimantis ridens) across the lowland wet forests of isthmian Central America. Mol Phylogenet Evol 47:992–1004
Wang IJ (2013) Examining the full effects of landscape heterogeneity on spatial genetic variation: A multiple matrix regression approach for quantifying geographic and ecological isolation. Evolution 67:3403–3411
Wang IJ, Bradburd GS (2014) Isolation by environment. Mol Ecol 23:5649–5662
Wang Y-H, Yang K-C, Bridgman CL, Lin L-K (2008b) Habitat suitability modelling to correlate gene flow with landscape connectivity. Landsc Ecol 23:989–1000
Webb SD (2006) The Great American Biotic Interchange: patterns and processes. Ann Mo Bot Gard 93:245–257
Weigt LA, Crawford AJ, Rand AS, Ryan MJ (2005) Biogeography of the túngara frog, Physalaemus pustulosus: a molecular perspective. Mol Ecol 14:3857–3876
Weir JT, Bermingham E, Schluter D (2009) The Great American biotic interchange in birds. Proc Natl Acad Sci USA 106:21737–21742
Werle E, Schneider C, Renner MV, W F (1994) Convenient single-step, one tube purification of PCR products for direct sequencing. Nucleic Acids Res 22:4354–4355
Weyl R (1980) Geology of Central America. Gebrueder Borntraeger, Berlin-Stuttgart
Wilson JS, Carril OM, Sipes SD (2014) Revisiting the Great American Biotic Interchange through analyses of amphitropical bees. Ecography 37:791–796
Wollenberg KC, Vieites DR, Glaw F, Vences M (2011) Speciation in little: the role of range and body size in the diversification of Malagasy mantellid frogs. BMC Evol Biol 11:217
Wollenberg Valero KC (2015) Evidence for an intrinsic factor promoting landscape genetic divergence in Madagascan leaf-litter frogs. Front Genet 6:155
Wright S (1943) Isolation by distance. Genetics 28:114–138
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Zhang J, Kapli P, Pavlidis P, Stamatakis A (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29:2869–2876
Zuur AF, Ieno EN, Elphick CS (2010) A protocol for data exploration to avoid common statistical problems. Methods Ecol Evol 1:3–14
Acknowledgements
We thank Federico Bolaños (UCR) for permission and access to the samples sequenced for this study and Sandra “Vicky” Flechas for her patient collaboration during lab procedures. Javier Guevara and the Sistema Nacional de Áreas de Conservación de Costa Rica provided collecting permits that ended up in most of the samples here analyzed. We wish to acknowledge funding from the Smithsonian Institution’s DNA Barcode Network (FY11 Award cycle to E. Bermingham) and the use of facilities at Naos Marine and Molecular Labs, Smithsonian Tropical Research Institute, Panama. AG-R is currently supported by a postdoctoral fellowship from Dirección General de Asuntos del Personal Académico (DGAPA) at Instituto de Biología, Universidad Nacional Autónoma de México. AG-R also acknowledges Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (CAPES) for the financial support during his PhD and Gerardo Chaves (MZUCR) for his teachings during innumerous field trips, as well as for the constant discussion on the herpetology of Central America. AJC was supported by Research Program INV-2017-51-1432 from the School of Sciences, Universidad de los Andes. Víctor Acosta and Brian Gratwicke kindly provided the photos used in Fig. 3. Fabricio Villalobos, Diogo Provete, Sergio Lima, Erick Arias-Piedra, and three anonymous reviewers provided valuable feedback to improve the quality of this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Barbara Mable
Supplementary information
Rights and permissions
About this article

Cite this article
García-Rodríguez, A., Guarnizo, C.E., Crawford, A.J. et al. Idiosyncratic responses to drivers of genetic differentiation in the complex landscapes of Isthmian Central America. Heredity 126, 251–265 (2021). https://doi.org/10.1038/s41437-020-00376-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-00376-8
This article is cited by
-
The roles of niche divergence, dispersal, and geology on the diversification of Neotropical true frogs from the Rana palmipes species group (Amphibia, Anura, Ranidae) during the Great American Biotic Interchange
Organisms Diversity & Evolution (2023)
-
Contrasting environmental drivers of genetic and phenotypic divergence in an Andean poison frog (Epipedobates anthonyi)
Heredity (2022)
