
Abstract
Massive fish introductions have taken place throughout much of the world, mostly over the last 70 years, and present a major threat to the genetic diversity of native fishes. Introductions have been reported for European Phoxinus, a ubiquitous small cyprinid that populates a wide variety of habitats. Species delineation in European Phoxinus has proven difficult with one reason being ranges of distribution that often traverse drainage boundaries. The present study combines recent samples with museum samples to better understand the current distribution of Phoxinus species and their distributions prior to the massive introductions of fishes in Europe, and to evaluate the use of museum specimens for species distribution studies. For these purposes, genetic lineages from sites collected prior to 1900 (n = 14), and between 1900 and 1950 (n = 8), were analysed using two mitochondrial and nuclear markers. Although possible fish introductions were detected, our results show that the distribution of genetic lineages of museum samples is comparable to that of the extant lineages of European Phoxinus present in those areas. These observations suggest that in the studied ranges the distribution of Phoxinus lineages has been driven by natural processes.
Similar content being viewed by others
Introduction
Fish introductions present, along with habitat loss, one of the main threats to genetic diversity of native fishes (Bruton 1995; Harrison and Stiassny 1999; Jug et al. 2005; Scott and Helfman 2001). It is known that the Romans undertook such introductions (Balon 2004), but, according to Crivelli (1995), 60% of introduced fish species have been stocked during the last 70 years. Reasons for introductions include sport and aquaculture (De Silva et al. 2006; Stanković et al. 2015), but also stocking of ornamental ponds (Padilla and Williams 2004; Ahnelt 2016) and biological control (Pyke 2008). Nevertheless, many introductions have occurred unintentionally, such as with ballast waters (Johansson et al. 2018), as by-products of stocking of commercial species, or from angling with live baits (Gozlan et al. 2010).
Minnows of the genus Phoxinus Rafinesque 1820 are ubiquitous small cyprinids that populate a wide variety of habitats throughout northern Eurasia. They are found in both still and running waters, including mountain streams, lowland rivers and lakes, spanning a range of altitudes and climatic zones (Banarescu 1992; Frost 1943; Kottelat and Freyhof 2007; Tack 1940). According to the latest revision of European Phoxinus (Palandačić et al. 2017), the genus contains ten valid species and at least eight additional well-resolved genetic lineages. Although for some lineages a species name is available (e.g., Phoxinus morella), not all lineages have been taxonomically formalised due to a lack of morphological data. Recently, a new species Phoxinus krkae was described based on a previously detected genetic lineage (Bogutskaya et al. 2019). In general, species delineation in European Phoxinus has proven difficult because of the phenotypic diversity present within the different genetic lineages (Collin and Fumagalli 2015; Ramler et al. 2017; Bogutskaya et al. 2019). Compounding the problem are the diverse distribution ranges of the Phoxinus species and lineages. While for the Leuciscinae subfamily (at present, at the family level Leuciscidae) distributions are usually limited to particular river drainages (e.g., Hrbek et al. 2004; Doadrio and Carmona 2004, Perea et al. 2010), in European Phoxinus the species and lineage distributions do not follow such zoogeographical patterns, often traversing drainages or even basin boundaries (Palandačić et al. 2015, 2017). Moreover, at some sampling sites, hybrids between different genetic lineages have been detected (Palandačić et al. 2015, 2017; Corral‐Lou et al. 2019). The origin of the different ranges of Phoxinus species can be attributed to anthropogenic translocations, as several species introductions have been reported in the literature. For example, Miro and Ventura (2015) report the spread of Phoxinus contemporary with trout introductions in Pyrenean lakes, while Museth et al. (2007) suggest their spread has been due to angling with live baits in Norway. In addition, Knebelsberger et al. (2015) conclude that minnows from the River Danube have been introduced into the River Rhine. Nevertheless, some of the observed species ranges might be natural (Palandačić et al. 2015).
Museum specimens have been used to address questions of taxonomy (Krajewski et al. 1997), phylogenetics (Cooper et al. 1992; Su et al. 1999), population genetics (Thomas et al. 1990), and biogeography (Alfaro et al. 2012), and to assess the impact of stocking of commercially important fishes (Nielsen et al. 1999, Hansen et al. 2009). However, working with museum samples is challenging, because of the degradation and fragmentation of the DNA (Wandeler et al. 2007; Zimmermann et al. 2008). In Phoxinus, museum specimens have been used for taxonomic revision of the genus in Europe (Palandačić et al. 2017) and for geometric morphometric analysis of habitat influence on body shape (Ramler et al. 2017). They have also been helpful in determining the ancestor among the four different genetic lineages currently present in the River Rhine (Knebelsberger et al. 2015). However, in those and most other studies using museum material (see Burrell et al. 2014 and the references within), only one locus (the barcoding section of cytochrome oxidase I, COI) was used.
The primary aim of the present study was to combine recent and museum samples (for definitions see below) to better understand the current distribution of Phoxinus species and lineages, and to compare their ranges prior to the massive introductions of fishes in Europe. A second aim was to evaluate the use of museum samples for such studies, i.e. to map historical species distributions. For these purposes, a combination of two mitochondrial sections (COI; and cytochrome b, cytb) were used to determine the genetic lineages of both museum and recent samples. Because hybridisation and introgression have been recorded (as reported above) in European Phoxinus, the nuclear recombination activating gene 1 (RAG1) was added to the dataset. However, as difficulties have previously been reported for amplification of RAG1 in museum samples, the internal transcribed spacer 1 (ITS1) was also included in the analysis.
Materials and methods
Mitochondrial DNA (mtDNA)
In the present study, the term ‘museum’ is applied to samples deposited in the various museums that were accessed (Museum of Natural History Berlin, Germany; Natural History Museum of Geneva, Switzerland; Natural History Museum Vienna, Austria) but which were collected prior to the year 2000, while the term ‘recent’ is applied to samples collected in contemporary Phoxinus studies, mostly after the year 2000. Altogether, 186 museum samples from 18 countries, and 48 recent samples from eight countries, were combined with the available sequences deposited in Genbank (Geiger et al. 2014; Knebelsberger et al. 2015; Palandačić et al. 2015, 2017; Ramler et al. 2017; Schönhuth et al. 2018; Denys and Manne 2019). DNA extraction and amplification followed protocols described in Palandačić et al. (2017), adhering to all requirements for working with museum material, including using UV-irradiated utensils, a clean room, negative extraction controls, among others. Following polymerase chain reaction (PCR), COI and cytb fragments were visually inspected, aligned with MEGA 6.0 (Tamura et al. 2011) and combined together to form single sequences. During this process, overlapping fragments were checked for congruity. The barcoding region of COI was fully recovered (650 bp), while for cytb fragments of length 473 or 590 bp were obtained, depending on the DNA quality. The program MEGA 6.0 (Tamura et al. 2011) was used for constructing simple neighbour-joining trees from the COI and cytb sequences with which the genetic lineages of museum samples were determined.
Because cytb was not available for representatives of all putative species and clades in European Phoxinus, a phylogenetic tree was calculated using COI sequences only. The tree was constructed from complete COI sequences using Bayesian inference implemented in BEAST 1.8.0 (Drummond et al. 2012) and the Maximum-Likelihood method in PhyML (Guindon et al. 2010) following protocols (including model selection, program settings and burn in) described in detail in Palandačić et al. (2017). In contrast to that last study, phylogenetic analysis was performed with an outgroup chosen based upon the phylogenetic study of Imoto et al. (2013). The species chosen for the outgroup were Rhynchocypris lagowskii (AP009147), Tribolodon hakonensis (AB626855, now in the genus Pseudaspius according to Imoto et al. 2013) and Oreoleuciscus potanini (AB626851).
Nuclear DNA (nDNA)
To assist determination of possible hybridisation and introgression events, two nuclear genes were added to the dataset. RAG1 has been used previously in Phoxinus phylogenetic and taxonomic studies (Palandačić et al. 2015), but without successful amplification from museum samples (Palandačić et al. 2017). Therefore, a second nuclear gene—ITS1—was added to the dataset. Because ITS1 is short (350 bp in Phoxinus) and exist in cells in many copies, it is a promising candidate for such amplification. RAG1 was amplified using the protocol of Palandačić et al. (2017), while ITS1 was amplified using primers ITS1F (Wyatt et al. 2006) and ITS3R (Palandačić et al. 2010, following the protocol therein). Sequencing was performed in both directions at LGC Genomics (Berlin, Germany) and Microsynth (Vienna, Austria).
RAG1 sequences displayed heterozygous positions, and their gametic phase was determined using Phase 2.1 (Stephens et al. 2001; Stephens and Scheet 2005), implemented in DnaSP 5.10 (Librado and Rozas 2009). In ITS1, many sequences had a clean start, but exhibited double or even triple peaks at one or another point in the sequence. Even though studies have shown (e.g., Bos et al. 2007; Harrigan et al. 2008) that the coalescent-based Bayesian algorithm implemented in Phase is a reliable alternative to cloning, ITS1 sequences exhibited a complicated structure, pointing to insertions or deletions (indels) and to the possibility of more than two haplotype variants per individual. Thus, 2−5 individuals from each species and each genetic lineage were cloned to resolve their gametic phase.
For cloning, PCR was repeated with the same ITS1 primers but with high fidelity PlatinumTM Taq DNA polymerase (Invitrogen). Initial denaturation, denaturation and extension followed Platinum Taq instructions, while for annealing, two temperatures were used: two cycles at 58 °C and 35 cycles at 52 °C. The PCR products were cleaned with enzymes Exo and Sap (Affymetrix), following the manufacturer’s protocol, and cloned with the TOPO-TA© cloning kit (Invitrogen). Six clones were chosen from each sample, purified with the Qiagen PCR purification kit and sequenced with M13 universal primers in both directions by Microsynth Austria.
After cloning, the redundant clones (with identical haplotypes) were removed from the alignment. However, as suspected, more than two different haplotypes were detected within a single sample (Table S1, supplementary material). According to Buckler et al. (1997) and Bailey et al. (2003), there are several ways to distinguish between pseudogenes and functional copies in ITS sequences. Here, pseudogenes were identified by examining the sequences of the flanking regions (30 bp long at the 5′ end and 82 bp long at the 3′ end), which are the highly conservative 18S and 5.8S genes, where no single nucleotide polymorphisms (SNPs) are expected. Thus, the sequences that exhibited SNPs in the flanking regions were excluded as possible pseudogenes. Subsequently, cloned sequences, homozygotes and simple heterozygotes (with one-step indels or wobbles, which could be resolved by Phase; see above) were aligned with MAFFT V 7.305 on XSEDE (Katoh and Toh 2008) the CIPRES Science Gateway (version 3.3; 197; Miller et al. 2010). The alignment was adjusted manually, and is reported in the supplementary material. Following the coding protocol described in FastGap (Borchsenius 2009), gaps were coded as nucleotides. However, FastGap only codes gaps as present or absent, regardless of the nucleotide in that state. If the nucleotide in a certain state is present, it codes the nucleotide as A and a gap as C. Nevertheless, in the alignment, there might be present a third state, which Fastgap ignores. Thus, the gaps were inspected visually and missing signs (−) changed to a nucleotide not present in that state. If, for example, the three possibilities in a chosen state were −, A or G, the missing sign would be changed to one of the remaining possible nucleotides (C or T), hence gathering maximum information from the dataset.
Finally, unrooted minimum-spanning networks were constructed from RAG1 and ITS1 sequences with the median-joining algorithm (Bandelt et al. 1999) implemented in Network 5.1 (www.fluxus-engineering.com) with default settings.
Results
mtDNA
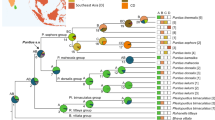
All museum and recent samples for which a genetic lineage was successfully determined (MEGA 6.0; Tamura et al. 2011) are reported in Table S1, including samples deposited in Genbank for which at least one mitochondrial gene and sample locality are available. For all the samples for which both genes (COI and cytb) are available (regardless of whether museum or recent), there were no incongruences in the genetic lineage detected. The main phylogenetic tree based on COI sequences showed good support for the clades, though it was unable to resolve the relationships among the lineages. Nevertheless, it confirmed the inclusion of museum samples in their respective clades and subclades (Fig. 1). The total number of samples for each lineage and the number of different haplotypes are reported in Table S2 (supplementary material). Based on these two analyses (lineage determination based on cytb and COI and phylogenetic analysis based on COI), 1190 samples from 268 localities were used to create a map showing the current distribution of Phoxinus lineages (Fig. 2). Besides the 18 clades reported previously in Palandačić et al. (2017), four new ones were also identified: two from the samples newly analysed in the present study (19, Kuban, Russia; and 20, Salgir, Ukraine), and one from a recently published study (21, East Pyrenees mountain range, Corral‐Lou et al. 2019). That last study was based upon variation at cytb and could not be included in the main COI phylogenetic tree. The fourth lineage (22, Ol'doy, Russia, Schönhuth et al. 2018) is also presented on the COI phylogenetic tree, though, because of its remoteness (eastern Russia) and because it is only one sample, it is absent from the map. In addition to new clades, three new subclades were also detected in clade 9 (9c, museum samples from Kolomyja, Ukraine; 9d, recent samples from Mures, Romania; and 9e, museum samples from Cluj, Romania). As seen from the previous studies, some clades and subclades represent species and potential species that are not taxonomically formalised. Thus, for a clearer understanding, the term genetic lineage will be used hereafter instead of the terms species, clade or subclade. Nevertheless, the species names are clearly depicted in all of the figures.

Phylogenetic tree constructed from the barcoding region of COI using Bayesian inference (BI) with BEAST 1.8.0 (Drummond et al. 2012). Branches carry posterior probabilities and bootstrap supports (BS) from the tree constructed with the Maximum-Likelihood (ML) method (PhyML; Guindon et al. 2010). The tree is shaded according to the value of posterior probabilities: the lighter the shade the weaker its support. Only posterior probabilities above 0.9 are shown. A lack of bootstraps originating from the difference between the BI and ML trees is denoted with −. Genetic lineages are presented in the diagram in the upper left corner. The genetic lineages, which are valid species, are written in black and the lineages, for which the species name is available but not valid, are in grey. For the genetic lineages, which were collected at a single sampling site, their locality is given. The outgroup consist of Rhynchocypris lagowskii (AP009147), Tribolodon hakonensis (AB626855) and Oreoleuciscus potanini (AB626851).

All studies are included. For clarity, subclades are annotated with letters, 1a–f, 5a–b, and 9a–e and black circles represent the approximate currently known distribution of subclades 1a–d. Major river drainages are shown.
Of 186 museum samples, the genetic lineage of 64 (34%) was successfully determined. Because the congruence of overlapping parts of adjacent fragments is an important quality control check, only samples with two or more successfully amplified overlapping fragments were used for further analysis. Amplification of all four COI fragments was successful in 53 museum samples, while in another four, the first three adjacent fragments were sequenced. In one sample, the last three fragments were successfully sequenced. Partial amplification of cytb was successful in 19 of 83 museum samples, of which six were not represented by COI (Table S1). Table S1 also includes details on the sampling sites, the GPS coordinates, and Genbank number and references where applicable.
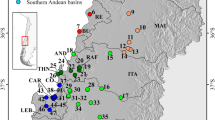
For clarity, the museum samples are presented in Fig. 3 in two groups: those collected before 1900 and those collected subsequently. All museum samples are shown and those originating from previous studies are denoted with their respective reference. The oldest sample for which the genetic lineage was successfully determined originates from the year 1836 (Austria, Palandačić et al. 2017). In addition, lineages of 14 samples collected before 1900, from Spain, France, Switzerland, Austria, Slovenia, Croatia, Serbia, Bulgaria, Ukraine and Germany (Knebelsberger et al. 2015), were successfully determined (Fig. 3a). The detected genetic lineages generally correspond to those that are currently present in the same areas (Fig. 3a, c). Moreover, some of the localities of the museum samples collected before 1900 and of the recent samples are (almost) the same, as are those of the lineages detected at the following sites: (i) Spain in 1864 and 1869; (ii) Austria in 1836 and 1889 (Ramler et al. 2017); (iii) Slovenia in 1892 and 1899; (iv) and Croatia in 1850 and 1897. However, when comparing the recent samples with the lineages detected in the samples collected in Germany in 1883 and 1888 (Knebelsberger et al. 2015), in Austria in 1842, and in Croatia in 1865, additional lineages were detected in the recent samples (marked with black arrows in Fig. 3c). In the three samples collected in Austria in 1877, two were assigned to genetic lineage 9a (P. marsilii) and one to lineage 5b. For other museum sampling sites (Switzerland, 1866; Serbia, 1869; Ukraine, 1883; and Bulgaria, 1894), comparative recent material from those sites or in their vicinity was not available.

This presents a closer view of Fig. 2, showing only the part of Europe for which the museum material is available. Genetic lineages were determined based on the mitochondrial genes COI and cytb. Lineages and attributed (currently valid) species are presented in the legend. Year of collection is noted beside each sampling site. Previous studies: K, Knebelsberger et al. 2015; P, Palandačić et al. 2017; R, Ramler et al. 2017. a Museum samples collected prior to 1900. b Museum samples collected after 1900. c All samples; museum samples denoted with a crosshair; arrows denote sample sites where fish introductions are identified.
Figure 3b shows the locations of samples collected between 1900 and 2000. Those collected in Germany in 1981, Austria in 1953, 1963, 1980 (two samples) and 1986, and Croatia in 1982 and 1986 were subsequent to the time of massive fish introductions and are, from this respect, equivalent to recent lineages. Samples from Croatia and Austria also exhibit the same genetic lineages as the recent samples, except for those collected in Austria in 1980, which belong to the lineage otherwise present in central Slovenia (1c, light green), and samples collected in 1986, which belong to lineage 1d. This lineage is distributed in Austria also in the recent samples and in the samples collected in 1925, but further south. The samples collected in Germany in 1981 were classified to genetic lineage 11. However, no recent samples are available for comparison. There are also no recent samples for comparison with the one collected in Poland in 1921 (lineage 9a), the sample collected in Ukraine in 1900 (9c; Ramler et al. 2017), and the one in Romania in 1902 (9e). Samples collected in Montenegro in 1917 (Palandačić et al. 2017) were classified as lineage 5a together with the recent samples from that area, though no recent data from the proximity are available. Meanwhile, the samples collected in Bosnia-Herzegovina in 1906 (lineage 1e) correspond to the recent ones from Serbia (Fig. 3c), where both clades 1e and 5a are present.
At two sites, sampling has been undertaken several times over the last 200 years. In Vienna, samples were collected in 1836, 1909, 1953 and 2014, and all exhibited the same haplotype (lineage 9a). Krk Island, Croatia, was sampled in 1850, 1982, 1986 and 2000, the lineage detected from which was 1a, and has not changed, though the haplotypes were not the same. In three samples from central Germany (Durkheim, collected in 1838), the complete COI was amplified successfully. However, the samples clustered into a clade together with Lake Ohrid (North Macedonia) samples. The same result was observed when amplifying cytb and ITS1, indicating a mistake in the labelling; these samples were thus excluded from the analysis.
nDNA
For the gametic phase determination, 97 1258-bp-long RAG1 sequences were used, 55 of which were new, and 107 downloaded from Genbank. However, some of the Genbank sequences (Schönhuth et al. 2018) included unknown nucleotide bases (N), and were thus omitted from further analysis. In addition, some of the calculated haplotypes exhibited low probabilities for several heterozygous positions and were also excluded. Finally, only the haplotypes that had no ambiguous sites, and no more than one heterozygous position determined with a probability of less than 0.9, were included in the network construction. Altogether, the median-joining network was constructed from 140 sequences (280 resolved haplotypes) representing 27 of 33 genetic lineages known in Phoxinus. The RAG1 network (Fig. 4a) presented an unclear structure, with the lineages based upon mtDNA forming few identifiable groups (lineages 3, 4, 6, 7 + 8, 14, 15 and 18). The haplotypes of other well-resolved mtDNA lineages are scattered across the network, with the haplotypes from samples as distant from each other as lineages 13 (Iberian Peninsula) and 19 (Kuban River, Russia) clustering together. The newly detected lineages, first reported in the present paper, are marked with arrows.

a Haplotype network constructed with RAG1. Colours represent lineages detected by mtDNA analysis, and are shown in the legend. The gametic phase of heterozygous individuals was determined using Phase 2.1 (Stephens et al. 2001, Stephens and Scheet 2005). An unrooted minimum-spanning network was constructed with the median-joining algorithm (Bandelt et al. 1999) implemented in Network 5.1 (www.fluxus-engineering.com) with default settings. The lines carry the number of mutations where more than one. Circle size corresponds to haplotype frequency, with the biggest encompassing 28 samples. Arrows denote the lineages first presented by RAG1. b ITS1 haplotype network constructed using homozygotes, simple heterozygotes resolved by Phase 2.1 (Stephens et al. 2001, Stephens and Scheet 2005), and cloned samples. Lines carry the number of mutations when more than one. Circle size corresponds to haplotype frequency, with the biggest encompassing 105 samples. However, sampling was not distributed equally among the lineages, cloning revealed more than two haplotypes per sample and did not have the same success rate in all the lineages, while allele dropout might be present in museum samples. Thus, the size of the circles do not project a realistic picture of haplotype frequencies.
The ITS1 dataset included 290 cloned sequences, homozygotes and simple heterozygotes, representing 13 of the Phoxinus genetic lineages. Of those, 23 samples of lineages 1a, 2, 3, 5a, 6, 7, 8 and 9a were successfully cloned (Table S1), while clades 4, 11, 12, 13 and 14 were represented by homozygotes and simple heterozygotes (with cloning unsuccessful). Cloning of museum samples failed, though homozygotes and simple heterozygotes were successfully sequenced. In the ITS1 network, the lineage grouping is familiar and confirms the lineages detected by mtDNA. However, due to technical problems (unsuccessful cloning, insufficient pseudogene designation, gap coding), the results for that network are not very reliable.
Discussion
The distribution of species in the genus Phoxinus does not correspond to river drainages or even sea basins, an unusual pattern for leusciscins (Gómez and Lunt 2007; Perea et al. 2010; but see Levin et al. 2017). Thus, the question arises of whether this distribution is natural or anthropogenic in origin. According to Museth et al. (2007), human-assisted introduction of Phoxinus species in Norway had already taken place in the late nineteenth century, mostly in the southern part of the country, with translocations becoming more frequent after 1950. Miro and Ventura (2015) report that the first introductions of Phoxinus in the Pyrenees took place after 1970. In the present study, the genetic lineage of 14 samples collected prior to 1900 and eight samples collected between 1900 and 1950 (Fig. 3a, b) were analysed, and the distribution of the detected lineages is comparable to those present in the same areas today. Moreover, in the samples collected in Austria in 1877, two bordering lineages (5b and 9a) were detected, pointing to a natural co-occurrence of two lineages. Nevertheless, as detected in Germany (Knebelsberger et al. 2015), the results point to fish introductions also in Austria and, possibly, Croatia. For some of the museum samples, comparison with recent samples was not possible. However, their inclusion helps expand the knowledge on some of the species ranges. Thus, combining museum and recent material proves to be a good strategy for better understanding species ranges, especially in genera where fish introductions are common.
Understanding the distribution of European Phoxinus using a combination of museum and recent material
In the last two decades, considerable efforts have been made to inventorise the number of genetic lineages comprising the European P. phoxinus species complex (Kottelat and Freyhof 2007; Bianco 2014; Palandačić et al. 2015, 2017; Corral‐Lou et al. 2019, Bogutskaya et al. 2019). Even so, the exact number remains unknown and their taxonomic formalisation incomplete. In the present study, museum material has helped to confirm the distribution areas of some Phoxinus genetic lineages that are probably a consequence of natural processes, and highlight the areas where fish introductions have taken place.
In the Balkan Peninsula, an unusual distribution of Phoxinus lineages has been reported previously (Palandačić et al. 2015, 2017), attributed to migration of fishes through underground water connections characteristic of karst landscapes, reported also in other studies (Borowsky and Mertz 2001; Dillman et al. 2010; Palandačić et al. 2012). Yet, the pattern being a consequence of stocking could not be excluded. Here, museum material collected in Serbia (1869), Bosnia-Hercegovina (1906) and Montenegro (1917) seems to confirm the patchy distribution of lineages 1e (dark green) and 5a (red) in the central Balkans area even before most of the massive introductions took place. The results appear to support the hypothesis that the unique species distributions of Phoxinus in the karst areas are a result of natural processes (Palandačić et al. 2015).
On a larger scale, the museum samples used in the present study confirm the distribution of seven genetic lineages (1d, 1e, 5a, 5b, 9a, 9c, 9d) in the Danube drainage, where at least 17 lineages (Table S1: 1a–f, 2, 3, 4, 5a–b, 9a–e and 11) have been detected previously. These lineages are distributed discretely in the upper, the middle, and the lower Danube, and in left and right tributaries (Figs. 2 and 3). Museum samples confirm the distribution of lineage 13 from north-west Spain to south-west France, in congruence with earlier studies (Kottelat 2007; Geiger et al. 2014). Moreover, the samples collected in Pau, France, in 1869 are in close proximity to (about 50 km from) the type locality of P. bigerri (lineage 13; Adour River in Tarbes), supporting its historical distribution in this area. In addition to museum material, recent samples provide further information on the ranges of Phoxinus, mostly for lineage 9, which is distributed in the left tributaries of the middle and lower Danube, and lineage 17, which seems to be spread across northern Europe all the way to the Ural Mountains. Besides adding new data on the distribution of previously recognised lineages, two new lineages (19 and 20) were detected among the recent samples included in the present study. Lineage 19 is located in Kuban, Russia, while lineage 20 is from Salgir, Crimea. For the latter, a species name is available, P. chrysoprasius (Pallas 1814). However, the lineage is not well supported by the RAG1 network. In contrast, lineage 9d, for which a name is also available (P. carpathicus Popescu Gorji and Dimitriu 1950; type locality, Lake Rosu (Red Lake), Romania), is slightly supported in the RAG1 network (Fig. 4a).
While the threat of fish introductions is more pronounced with commercially interesting fish species (Sušnik et al. 2004; Jug et al. 2005), the co-occurrence of minnows with salmonids caused the introduction of the former across much of Europe (Kottelat 2007; Kottelat and Freyhof 2007; Museth et al. 2007). Furthermore, the Rhine–Main–Danube canal and the Rhone–Rhine canal connect together most of the biggest river drainages on the continent. In congruence with this, Knebelsberger et al. (2015) found four different Phoxinus species or clades in the River Rhine, with some introduced from the Danube. Meanwhile, Corral‐Lou et al. (2019; see also the references therein) report the introduction of P. septimaniae in north-east Spain, while, in Croatia, mixed haplotypes in one Adriatic river have been attributed to water exchange serving a reversible power plant (Vučić et al. 2018). In general, introductions of Phoxinus are well recorded (Aparicio et al. 2000; Kottelat 2007; Kottelat and Freyhof 2007; Museth et al. 2007; Schreiber and Sosat 2007; Benejam et al. 2010; Maceda‐Veiga et al. 2010; Knebelsberger et al. 2015; Miro and Ventura 2015), but, in contrast, only two cases of fish introduction are suggested here (lineages 1a and 10 in Austria, and lineage 1a in Croatia), pointing to a lesser effect of fish introductions on European Phoxinus than reported in the previous studies. Nevertheless, these results could be influenced by the limitations of museum material (e.g., number of samples analysed, see section ‘Usefulness of museum samples in species distribution studies’ and Table S1). Thus, conservation of Phoxinus autochthonous populations should be taken seriously, especially because detailed studies of the genus throughout Europe are still missing. As seen in the present and previous studies, every time new data are reported, new genetic lineages of European Phoxinus emerge. In addition, the use and interpretation of nuclear markers has been challenging, with most studies (including the present one, see the section ‘Some comments on nuclear markers used’) relying on mitochondrial data. Thus, more studies, mostly concerning northern European discharges, are needed to finalise species delimitation and form conservation guidelines. Meanwhile, fish management should be more environmentally protective. Supplemental stocking (if needed at all) should be conducted with care, and introduction of non-target (or by-product) species should be prevented.
Usefulness of museum samples in species distribution studies
The present study confirms the usefulness of museum material (some of which is more than 180 years old) to gain a better understanding of species distributions, especially in cases where fish introductions have been common. Nevertheless, using museum material does present challenges. In large, old museum collections, such as at the Natural History Museum Vienna, mislabelling of specimens can occur (previously reported for this collection by Bogutskaya and Zupančič 1999) and was also detected in the present study. The population that, according to the museum label, originated from central Germany clustered with Lake Ohrid samples based upon analysis of three different genes, indicating that the label information needs to be considered with caution (see also Boessenkool et al. 2010; Rawlence et al. 2014; Paterson et al. 2016). Further, successful amplification of fragments depends upon the preservation methods used for museum samples (Hall et al. 1997), while the exact method is usually unknown. In fishes, conservation methods can be distinguished, as alcohol-fixed specimens possess white eyes, while formalin-fixed fishes have clear black eyes (De Bruyn et al. 2011). Here, most (175 out of 186) of the specimens analysed were alcohol-fixed, yet, the genetic lineage was successfully determined in only one-third of individuals. Finally, degradation and fragmentation of DNA—resulting in a low success rate despite a large amount of work—is the reason that most studies use only a few loci when analysing museum samples (Burrell et al. 2014). This is especially true for nuclear loci, as they are typically less variable than mtDNA and longer stretches of DNA are needed for the analysis. Correspondingly, analysis of nuclear loci was the most challenging in the present study.
Some comments on nuclear markers used
Because of several cases of hybridisation or introgression, informative nuclear markers are crucial for studying the phylogeny of the P. phoxinus species complex. As found previously (Palandačić et al. 2017), RAG1 could not be amplified in museum samples. Moreover, it showed only limited resolution in species delimitation investigations (Palandačić et al. 2015; Corral‐Lou et al. 2019). In the present study, RAG1 analysis was challenging (see Results), and the network that was constructed was weakly resolved, with many cross-connections among the haplotypes (Fig. 4a). Most notably, despite inclusion of more samples, representing almost all the genetic lineages detected with mtDNA analysis (27 of 33), no more information became available, with haplotypes from otherwise distant and well-resolved lineages scattered across the network (see, e.g., lineages 13 and 17 in Fig. 4). Due to the problems with RAG1 analysis, it is hard to say whether these incongruities are a consequence of artefacts of the sequencing or gametic phase determination, or of the lack of signal, or both. The second nuclear marker—ITS1—seems more promising as it is short (350 bp) and sequencing from museum material was successful. In addition, it has previously been used for barcoding in fungi and plants (Schoch et al. 2012; Cheng et al. 2016) and for testing for hybridisation in fishes (Wyatt et al. 2006; Hamilton and Tyler 2007). Indeed, in comparison to the RAG1 network, the ITS1 network presented better resolution (support) for the genetic lineages detected with mtDNA (Fig. 4b), especially when large numbers of samples were sequenced (e.g., lineage 1a). However, several outliers (possible pseudogenes in, e.g., lineage 13) pointed to unsuccessful amplification or detection, or both, of functional ITS1 copies. Thus, the weaknesses with the use of ITS1 as a marker, resulting from concerted evolution, unequal crossing-over, and existence of pseudogenes and multiple paralogues (Arnheim 1983; Bayly and Ladiges 2007) are apparent. It was not an aim of the present study to evaluate RAG1 and ITS1 as markers, but it can be deduced that neither is appropriate for species delimitation and both have limited power for detecting hybrids in European Phoxinus.
With new techniques on the rise, the limitations of using PCR-based methods on degraded museum material might be overcome by using DNA capture hybridisation and whole genome sequencing methods (Bailey et al. 2016; Souza et al. 2017), thus enabling the use of many nuclear markers and whole mitochondrial genomes (Mason et al. 2011). As museum specimens are unique biological material and their existence represents a finite resource, sampling is better justified with material being analysed with the latest (and thus most promising) techniques. Nevertheless, using those methods for extracting molecular data in a cost-effective way may still represents an obstacle (Knyshov et al. 2019). Thus, this study shows that even with Sanger sequencing and a limited number of markers, a combination of museum and recent material can provide valuable insights for the conservation of fishes.
Data archiving
All sequences are available under Genbank accession numbers MN820726−MN820819, MN816034−MN816147, MN818456−MN818521, MN818001−MN818455. In addition, the datasets supporting this article have been uploaded as supplementary material (Table S1, ITS1 aligned fasta file).
References
Ahnelt H (2016) Translocations of tropical and subtropical marine fish species into the Mediterranean. A case study based on Siganus virgatus (Teleostei: Siganidae). Biologia 71:952–959. https://doi.org/10.1515/biolog-2016-0106
Alfaro JWL, Boubli JP, Olson LE, Di Fiore AD, Wilson B, Gutierrez-Espeleta GA et al. (2012) Explosive Pleistocene range expansion leads to widespread Amazonian sympatry between robust and gracile capuchin monkeys. J Biogeogr 39:272–288. https://doi.org/10.1111/j.1365-2699.2011.02609.x
Aparicio E, Vargas MJ, Olmo JM, de Sostoa A (2000) Decline of native freshwater fishes in a Mediterranean watershed on the Iberian Peninsula: a quantitative assessment. Environ Biol Fish 59(1):11–19
Arnheim N (1983) Concerted evolution in multigene families. In: Nei M, Koehn R (eds) Evolution of genes and proteins. Sinauer, Sunderland, MA, p 38–61
Bailey CD, Carr TG, Harris SA, Hughes CE (2003) Characterization of angiosperm nrDNA polymorphism, paralogy, and pseudogenes. Mol Phylogenet Evol 29:435–455. https://doi.org/10.1016/j.ympev.2003.08.021
Bailey SE, Mao X, Struebig M, Tsagkogeorga G, Csorba G, Heaney LR et al. (2016) The use of museum samples for large-scale sequence capture: a study of congeneric horseshoe bats (family Rhinolophidae). Biol J Linn Soc 117:58–70. https://doi.org/10.1111/bij.12620
Balon EK (2004) About the oldest domesticates among fishes. J Fish Biol 65(s1):1–27. https://doi.org/10.1111/j.0022-1112.2004.00563.x
Banarescu P (1992) Zoogeography of fresh waters. Vol. II. Distribution and dispersal of freshwater animals in North America and Eurasia. AULA-Verlag, Wiesbaden, Germany
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16(1):37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Bayly MJ, Ladiges PY (2007) Divergent paralogues of ribosomal DNA in eucalypts (Myrtaceae). Mol Phylogen Evol 44:346–356. https://doi.org/10.1016/j.ympev.2006.10.027
Benejam L, Angermeier PL, Munne A, García‐Berthou E (2010) Assessing effects of water abstraction on fish assemblages in Mediterranean streams. Freshw Biol 55(3):628–642. https://doi.org/10.1111/j.1365-2427.2009.02299.x
Bianco PG (2014) An update on the status of native and exotic freshwater fishes of Italy. J Appl Ichthyol 30:62–77. https://doi.org/10.1111/jai.12291
Boessenkool S, Star B, Scofiled RP, Seddon PJ, Walters JM (2010) Lost in translation or deliberate falsification? Genetic analyses reveal erroneous museum data for historic penguin specimens. Proc R Soc B 277:1057–1064. https://doi.org/10.1098/rspb.2009.1837
Bogutskaya NG, Zupančič P (1999) A re-description of Leuciscus zrmanjae (Karaman, 1928) and new data on the taxonomy of Leuciscus illiricus, L. svallize and L. cephalus (Pisces: Cyprinidae) in the West Balkans. Ann Naturhist Mus Wien 101B:509–529
Bogutskaya NG, Jelić D, Vucić M, Jelić M, Diripasko OA, Stefanov T, et al. (2019) Description of a new species of Phoxinus from the upper Krka River (Adriatic Basin) in Croatia (Actinopterygii: Leuciscidae), first discovered as a molecular clade. J Fish Biol. https://doi.org/10.1111/jfb.14210
Borchsenius F (2009) FastGap 1.2. Department of Biosciences, Aarhus University, Denmark. Published online at http://www.aubot.dk/FastGap_home.htm
Borowsky RB, Mertz L (2001) Genetic differentiation among populations of the cave fish Schistura oedipus (Cypriniformes: Balitoridae). Environ Biol Fishes 62:225–231
Bos DH, Turner SM, Andrew DeWoody J (2007) Haplotype inference from diploid sequence data: evaluating performance using non-neutral MHC sequences. Hereditas 144(6):228–234
Bruton MN (1995) Have fishes had their chips? The dilemma of threatened fishes. Environ Biol Fishes 43:1–27
Buckler ES, Ippolito A, Holtsford TP (1997) The evolution of ribosomal DNA: divergent paralogues and phylogenetic implications. Genetics 145:821–832
Burrell AS, Disotell TR, Bergey CM (2014) The use of museum specimens with high-throughput DNA sequencers. J Hum Evol 79:35–44. https://doi.org/10.1016/j.jhevol.2014.10.015
Cheng T, Xu C, Lei L, Li C, Zhang Y, Zhou S (2016) Barcoding the kingdom Plantae: new PCR primers for ITS regions of plants with improved universality and specificity. Mol Ecol Resour 16:138–149. https://doi.org/10.1111/1755-0998.12438
Collin H, Fumagalli L (2015) The role of geography and ecology in shaping repeated patterns of morphological and genetic differentiation between European minnows (Phoxinus phoxinus) from the Pyrenees and the Alps. Biol J Linn Soc 116:691–703
Cooper A, Mourer-Chauviré C, Chambers GK, von Haeseler A, Wilson AC, Pääbo S (1992) Independent origins of New Zealand moas and kiwis. Proc Natl Acad Sci 89:8741–8744. https://doi.org/10.1073/pnas.89.18.8741
Corral‐Lou A, Perea S, Aparicio E, Doadrio I (2019) Phylogeography and species delineation of the genus Phoxinus Rafinesque, 1820 (Actinopterygii: Leuciscidae) in the Iberian Peninsula. J Zool Syst Evol Res 00:1–16. https://doi.org/10.1111/jzs.12320
Crivelli AJ (1995) Are fish introductions a threat to endemic freshwater fishes in northern Mediterranean region? Biol Cons 72:311–319. https://doi.org/10.1016/0006-3207(94)00092-5
De Bruyn M, Parenti LR, Carvalho GR (2011) Successful extraction of DNA from archived alcohol-fixed white-eye fish specimens using an ancient DNA protocol. J Fish Biol 78:2074–2079. https://doi.org/10.1111/j.1095-8649.2011.02975.x
Denys GPJ, Manne S (2019) First record of Phoxinus csikii Hankó, 1922 (Actinopterygii, Cypriniformes) in France. Société Française d'Ichtyologie. Cybium 43(2):199–202
De Silva SS, Nguyen TTT, Abery NW, Amarasinghe US (2006) An evaluation of the role and impacts of alien finfish in Asian inland aquaculture. Aquacult Res 37:1–17. https://doi.org/10.1111/j.1365-2109.2005.01369.x
Dillman CB, Bergstrom DE, Noltie DB, Holtsford TP, Mayden RL (2010) Regressive progression, progressive regression or neither? Phylogeny and evolution of the Percopsiformes (Teleostei, Paracanthopterygii). Zool Scr 40(1):45–60
Doadrio I, Carmona JA (2004) Phylogenetic relationships and biogeography of the genus Chondrostoma inferred from mitochondrial DNA sequences. Mol Phyl Evol 33(3):802–815. https://doi.org/10.1016/j.ympev.2004.07.008
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29(8):1969–1973. https://doi.org/10.1093/molbev/mss075
Frost WE (1943) The natural history of the minnow, Phoxinus phoxinus. J Anim Ecol 12:139–162
Geiger MF, Herder F, Monaghan MT, Almada V, Barbieri R, Bariche M et al. (2014) Spatial heterogeneity in the Mediterranean Biodiversity Hotspot affects barcoding accuracy of its freshwater fishes. Mol. Ecol Res 14:1210–1221
Gómez A, Lunt D (2007) Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula. In: Weiss S, Ferrand N (eds) Phylogeography of Southern European refugia. Springer, Dordrecht, p 155−188
Gozlan RE, Britton JR, Cowx IG, Copp GH (2010) Current knowledge on non-native freshwater fish introductions. J Fish Biol 76:751–786. https://doi.org/10.1111/j.1095-8649.2010.02566.x
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate Maximum-Likelihood Phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321. https://doi.org/10.1093/sysbio/syq010
Hall LM, Willcox MS, Jones DS (1997) Association of enzyme inhibition with methods of museum skin preparation. Biotechniques 22:928–934
Hamilton PB, Tyler CR (2007) Identification of microsatellite loci for parentage analysis in roach Rutilus rutilus and eight other cyprinid fish by cross-species amplification, and a novel test for detecting hybrids between roach and other cyprinids. Mol Ecol Notes 8:462–465. https://doi.org/10.1111/j.1471-8286.2007.01994.x
Hansen MM, Fraser DJ, Meier K, Mensberg KLD (2009) Sixty years of anthropogenic pressure: a spatio-temporal genetic analysis of brown trout populations subject to stocking and population declines. Mol Ecol 18:2549–2562. https://doi.org/10.1111/j.1365-294X.2009.04198.x
Harrigan RJ, Mazza ME, Sorenson MD (2008) Computation vs. cloning: evaluation of two methods for haplotype determination. Mol Ecol Resour 8(6):1239–1248. https://doi.org/10.1111/j.1755-0998.2008.02241.x
Harrison IJ, Stiassny MLJ (1999) The quiet crisis: a preliminary listing of the freshwater fishes of the world that are extinct or “missing in action”. In: MacPhee RDE (ed) Extinctions in near time. Kluwer Academic/Plenum, New York, NY, p 271–331
Hrbek T, Stölting KN, Bardakci F, Küçük F, Wildekamp RH, Meyer A (2004) Plate tectonics and biogeographical patterns of the Pseudophoxinus (Pisces: Cypriniformes) species complex of central Anatolia, Turkey. Mol Phylog Evol 32:297–308. https://doi.org/10.1016/j.ympev.2003.12.017
Imoto JM, Saitoh K, Sasaki T, Yonezawa T, Adachi J, Kartavtsev YP et al. (2013) Phylogeny and biogeography of highly diverged freshwater fish species (Leusciscinae, Cyprinidae, Teleostei) inferred from mitochondrial genome analysis. Gene 514:112–124. https://doi.org/10.1016/j.gene.2012.10.019
Johansson ML, Dufour BA, Wellband KW, Corkum LD, MacIsaac HJ, Heath DD (2018) Human-mediated and natural dispersal of an invasive fish in the eastern Great Lakes. Heredity 120:533–546
Jug T, Berrebi P, Snoj A (2005) Distribution of non-native trout in Slovenia and their introgression with native trout populations as observed through microsatellite DNA analysis. Biol Cons 123:381–388. https://doi.org/10.1016/j.biocon.2004.11.022
Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9:286–298. https://doi.org/10.1093/bib/bbn013
Knebelsberger T, Dunz AR, Neumann D, Geiger MF (2015) Molecular diversity of Germany's freshwater fishes and lampreys assessed by DNA barcoding. Mol Ecol Resour 15(3):562–572. https://doi.org/10.1111/1755-0998.12322
Knyshov A, Gordon ERL, Weirauch C (2019) Cost‐efficient high throughput capture of museum arthropod specimen DNA using PCR-generated baits. Methods Ecol Evol 10 (6) https://doi.org/10.1111/2041-210X.13169
Kottelat M (2007) Three new species of Phoxinus from Greece and southern France (Teleostei: Cyprinidae). Ichthyol Explor Freshw 18:145–162
Kottelat M, Freyhof J (2007) Handbook of European Freshwater Fishes. Steven Simpson Books, Berlin
Krajewski C, Buckley L, Westerman M (1997) DNA phylogeny of the marsupial wolf resolved. Proc R Soc Lond B Biol Sci 264:911–917. https://doi.org/10.1098/rspb.1997.0126
Levin BA, Simonov EP, Ermakov OA, Levina MA, Interesova EA, Kovalchuk OM et al. (2017) Phylogeny and phylogeography of the roaches, genus Rutilus (Cyprinidae), at the Eastern part of its range as inferred from mtDNA analysis. Hydrobiologia 788(1):33–46. https://doi.org/10.1007/s10750-016-2984-3
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Maceda‐Veiga A, Monleon‐Getino A, Caiola N, Casals F, de Sostoa A (2010) Changes in fish assemblages in catchments in north‐eastern Spain: biodiversity, conservation status and introduced species. Freshw Biol 55(8):1734–1746. https://doi.org/10.1111/j.1365-2427.2010.02407.x
Mason VC, Li G, Helgen KM, Murphy WJ (2011) Efficient cross-species capture hybridization and next-generation sequencing of mitochondrial genomes from noninvasively sampled museum specimens. Genome Res 21(10):1695–1704. https://doi.org/10.1101/gr.120196.111
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), The Institute of Electrical and Electronics Engineers, New Orleans, p 1–8
Miro A, Ventura M (2015) Evidence of exotic trout mediated minnow invasion in Pyrenean high mountain lakes. Biol Invasions 17:791–803. https://doi.org/10.1007/s10530-014-0769-z
Museth J, Hesthagen T, Sandlund OT, Thorstad EB, Ugedal O (2007) The history of the minnow Phoxinus phoxinus (L.) in Norway: from harmless species to pest. J Fish Biol 71(Suppl D):184–195. https://doi.org/10.1111/j.1095-8649.2007.01673.x
Nielsen EE, Hansen MM, Loeschcke V (1999) Analysis of DNA from old scale samples: technical aspects, applications and perspectives for conservation. Hereditas 130:265–276. https://doi.org/10.1111/j.1601-5223.1999.00265.x
Padilla DK, Williams SL (2004) Beyond ballast water: Aquarium and ornamental trades as sources of invasive species in aquatic ecosystems. Front Ecol Environ 2:131–138. https://doi.org/10.1890/1540-9295(2004)002[0131:BBWAAO]2.0.CO;2
Palandačić A, Bravničar J, Zupančič P, Šanda R, Snoj A (2015) Molecular data suggest a multispecies complex of Phoxinus (Cyprinidae) in the Western Balkan Peninsula. Mol Phylogen Evol 92:118–123. https://doi.org/10.1016/j.ympev.2015.05.024
Palandačić A, Matschiner M, Zupančič P, Snoj A (2012) Fish migrate underground: the example of Delminichthys adspersus (Cyprinidae). Mol Ecol 21(7):1658–1671. https://doi.org/10.1111/j.1365-294X.2012.05507.x
Palandačić A, Naseka A, Ramler D, Ahnelt H (2017) Contrasting morphology with molecular data: an approach to revision of species complexes based on the example of European Phoxinus (Cyprinidae). BMC Evol Biol 17:184. https://doi.org/10.1186/s12862-017-1032-x
Palandačić A, Zupančič P, Snoj A (2010) Revised classification of former genus Phoxinellus using nuclear DNA sequences. Biochem Sys Ecol 38:1069–1073. https://doi.org/10.1016/j.bse.2010.09.021
Pallas PS (1814) Zoographia Rosso-Asiatica, sistens omnium animalium in extenso Imperio Rossico et adjacentibus maribus observatorumrecensionem, domicilia, mores et descriptiones anatomen atque icones plurimorum. 3 vols. [1811–1814]. Academia Scientiarum, Petropolis ([SanktPetersburg)
Paterson G, Albuquerque S, Blagoderov V, Brooks S, Cafferty S, Cane E, et al. (2016) iCollections—digitising the British and Irish butterflies in the Natural History Museum, London. Biodivers Data J 1–16, https://doi.org/10.3897/BDJ.4.e9559
Perea S, Böhme M, Zupančič P, Freyhof J, Šanda R, Özuluğ M et al. (2010) Phylogenetic relationships and biogeographical patterns in circum-Mediterranean subfamily Leuciscinae (Teleostei, Cyprinidae) inferred from both mitochondrial and nuclear data. BMC Evol Biol 10(1):265. https://doi.org/10.1186/1471.2148.10.265
Popescu Gorji A, Dimitriu M (1950) Observaţiuni piscicole la Lacul Roşu (Ciuc). Buletinul Institutului de Cercetari şi Proiectari Piscicole 9:81–99
Pyke GH (2008) Plague minnow or mosquitofish? A review of the biology and impacts of introduced Gambusia species. Annu Rev Ecol Evol Syst 39:171–191. https://doi.org/10.1146/annurev.ecolsys.39.110707.173451
Ramler D, Palandačić A, Delmastro GB, Wanzenböck J, Ahnelt H (2017) Morphological divergence of lake and stream Phoxinus of Northern Italy and the Danube basin based on geometric morphometric analysis. Ecol Evol 7(2):572–584. https://doi.org/10.1002/ece3.2648
Rawlence NJ, Kennedy M, Waters JM, Scofield RP (2014) Morphological and ancient DNA analyses reveal inaccurate labels on two of Buller's bird specimens. J R Soc NZ 44(4):163–169. https://doi.org/10.1080/03036758.2014.972962
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA et al. (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA 109:6241–6246. https://doi.org/10.1073/pnas.1117018109
Schönhuth S, Vukić J, Šanda R, Yang L, Mayden RL (2018) Phylogenetic relationships and classification of the Holarctic family Leuciscidae (Cypriniformes: Cyprinoidei). Mol Phylog Evol 127:781–799. https://doi.org/10.1016/j.ympev.2018.06.026
Schreiber A, Sosat R (2007) The genetic population structure of the Eurasian fine-scaled minnow, Phoxinus phoxinus (Linnaeus 1758), in the contact area of the upper Rhine and Danube rivers (southwestern Germany) (Osteichthyes, Cypriniformes, Cyprinidae). Senckenb Biol 87(2):195–211
Scott MC, Helfman GS (2001) Native invasions, homogenization, and the mismeasure of integrity of fish assemblages. Fisheries 26(11):6–15. https://doi.org/10.1577/1548-8446(2001)026<0006:NIHATM>2.0.CO;2
Souza CA, Murphy N, Villacorta-Rath C, Woodings LN, Ilyushkina I, Hernandez CE et al. (2017) Efficiency of ddRAD target enriched sequencing across spiny rock lobster species (Palinuridae: Jasus). Sci Rep 7:6781. https://doi.org/10.1038/s41598-017-06582-5
Stanković D, Crivelli AJ, Snoj A (2015) Rainbow trout in Europe: introduction, naturalization, and impacts. Rev Fish Sci Aquac 23:39–71. https://doi.org/10.1080/23308249.2015.1024825
Stephens M, Scheet P (2005) Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet 76(3):449–462. https://doi.org/10.1086/428594
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68(4):978–989. https://doi.org/10.1086/319501
Su B, Wang YX, Lan H, Wang W, Zhang Y (1999) Phylogenetic study of complete cytochrome b genes in musk deer (genus Moschus) using museum samples. Mol Phylogen Evol 12:241–249. https://doi.org/10.1006/mpev.1999.0616
Sušnik S, Berebi P, Dovč P, Hansen MM, Snoj A (2004) Genetic introgression between wild and stocked salmonids and the prospects for using molecular markers in population rehabilitation: the case of the Adriatic grayling (Thymallus thymallus L. 1785). Heredity 93:273–282. https://doi.org/10.1038/sj.hdy.6800500
Tack E (1940) Die Elritze (Phoxinus laevis Ag.): eine monographische Bearbeitung. Arch Hydrobiol 37:321–425
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. https://doi.org/10.1093/molbev/msr121
Thomas WK, Pääbo S, Villablanca FX, Wilson AC (1990) Spatial and temporal continuity of kangaroo rat populations shown by sequencing mitochondrial DNA from museum specimens. J Mol Evol 31(2):101–112. https://doi.org/10.1007/BF02109479
Vučić M, Jelić D, Žutinić P, Grandjean F, Jelić M (2018) Distribution of Eurasian minnows (Phoxinus: Cypriniformes) in the Western Balkans. Knowl Manag Aquat Ec 419:11. https://doi.org/10.1051/kmae/201705
Wandeler P, Hoeck PE, Keller LF (2007) Back to the future: museum specimens in population genetics. Trends Ecol Evol 22:634–642. https://doi.org/10.1016/j.tree.2007.08.017
Wyatt PMW, Pitts CS, Butlin RK (2006) A molecular approach to detect hybridization between bream Abramis brama, roach Rutilus rutilus and rudd Scardinius erythrophthalmus. J Fish Biol 69:52–71. https://doi.org/10.1111/j.1095-8649.2006.01104.x
Zimmermann J, Hajibabaei M, Blackburn DC, Hanken J, Cantin E, Posfai J et al. (2008) DNA damage in preserved specimens and tissue samples: a molecular assessment. Front Zool 5:18. https://doi.org/10.1186/1742-9994-5-18
Acknowledgements
We would like to thank Dr. Nina Bogutskaya for productive discussion on the matters raised here. We would also like to thank Dr. Stephan Koblmüller (Institute of Zoology, University of Graz, Austria), Johanna Kapp and Dr. Peter Bartsch (Museum of Natural History Berlin, Germany), and Dr. Sonia Fisch-Muller (Department for Herpetology and Ichthyology, Natural History Museum of Geneva, Switzerland) for providing recent and museum material of Phoxinus from their collections. We would also like to thank Dr. Matthias Geiger, Dr. Fabian Herder and Dr. Jorg Freyhof for supplying the recent Phoxinus material, and we appreciate the support provided by the Project FREDIE (www.fredie.eu) funded by the Joint Initiative for Research and Innovation (PAKT) programme of the Leibniz Association. We would also like to thank Dr. Iain F. Wilson for editing the text. Finally, we would like to thank the editor Dr. Bastiaan Star and the three anonymous reviewers for their comments.
Funding
This study was funded partially by grant H-275981/2016 awarded by Hochschuljubiläumsstiftung der Stadt Wien, Vienna, Austria.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Palandačić, A., Kruckenhauser, L., Ahnelt, H. et al. European minnows through time: museum collections aid genetic assessment of species introductions in freshwater fishes (Cyprinidae: Phoxinus species complex). Heredity 124, 410–422 (2020). https://doi.org/10.1038/s41437-019-0292-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0292-1
This article is cited by
-
Evolutionary body shape diversification of the endemic Cyprinoidei fishes from the Balkan’s Dinaric karst
Organisms Diversity & Evolution (2023)
-
Molecular analysis reveals multiple native and alien Phoxinus species (Leusciscidae) in the Netherlands and Belgium
Biological Invasions (2022)
-
Increased spatial resolution of sampling in the Carpathian basin helps to understand the phylogeny of central European stream-dwelling gudgeons
BMC Zoology (2021)
-
Species composition of introduced and natural minnow populations of the Phoxinus cryptic complex in the westernmost part of the Po River Basin (north Italy)
Biological Invasions (2021)
-
Origin and history of Phoxinus (Cyprinidae) introductions in the Douro Basin (Iberian Peninsula): an update inferred from genetic data
Biological Invasions (2020)
