
Abstract
Continental islands are useful models to explore the roles of shared historical factors in the evolution of sympatric species. However, China’s largest continental group of islands, the Zhoushan Archipelago, was neglected by most studies focusing on biodiversity hotspots. Here we investigated the phylogeographic patterns and the historical demography of two sympatric hemipteran insects (Geisha distinctissima and Megacopta cribraria), which shared historical factors in the Zhoushan Archipelago. The results based on mtDNA (COX1, COX2-COX3, and CYTB) and nDNA (28S and ITS2) showed that G. distinctissima diverged into three genetic lineages (L1–L3) ~8.9–13.7 thousand years ago (kya), which coincided with the period of island isolation. However, the three lineages exhibit no clear phylogeographic patterns for frequent asymmetrical gene flow (starting around 5 kya) from the mainland and adjacent islands to other distant islands due to subsequent human activities. In contrast, only one genetic lineage exists for M. cribraria, without any phylogeographic structures. The ancestral range in the mainland as well as in neighboring islands, together with the frequent asymmetrical gene flow of M. cribraria (from the mainland and neighboring islands to more distant islands) within the last 5000 years suggests that human activities may have lead to the colonization of this species in the Zhoushan Archipelago. The contrasting genetic structures indicate shared historical factors but independent evolutionary histories for the two sympatric species in the Zhoushan Archipelago. Our demographic analysis clearly showed that both species underwent population expansion before 5 kya during the post-LGM (Last Glacial Maximum), which indicates that the two species shared concordant historical demographies. This result suggests that the population size of the two species was affected similarly by the climatic oscillations of post-LGM in Eastern China. Together, our findings reveal that the two insect species in the Zhoushan Archipelago exhibit contrasting genetic structures despite concordant historical demographies, which provides an important framework for the exploration of the evolution patterns of sympatric species in the continental island.
Similar content being viewed by others


Introduction
Islands represent ideal model ecosystems to explore the evolutionary processes of organisms. As islands are geographically isolated and fragmented habitats, they have long been used to investigate the mechanisms of biodiversity, speciation, adaptive radiation, and long-distance dispersal over oceans (Cowie and Holland 2006; Weigelt and Kreft 2013). Numerous studies have investigated oceanic islands, as they were never connected to any landmass and thus initially presented empty niches (Paulay 1994; Duryea et al. 2015). In contrast to oceanic islands, continental islands were intermittently connected with neighboring continents by the fluctuation of sea levels during Pleistocene glaciation, especially the Last Glacial Maximum [LGM, ∼1.8–2.3 thousand years ago (kya)] during the late Pleistocene (Tsukada 1988). Phylogeographic studies have provided empirical evidence that several historical events (e.g., climatic oscillation and the rising of sea level) can provide insight into the geographical distribution and demographic history of many flora and fauna populations worldwide (Byun et al. 1997; Bittkau and Comes 2005; Huang and Lin 2011; Campagna et al. 2012; Yang et al. 2017). Comparative phylogeography for multiple species has shown that sympatric species can exhibit concordant phylogeographic patterns because of shared historical events (Lyons 2003; Luo et al. 2016). However, several studies have identified contrasting phylogeographic patterns in many sympatric species, including invertebrates and mammals (Taberlet et al. 1998; Ditchfield 2000; Michaux et al. 2005; Lejeusne et al. 2011). As a result of the discordance in phylogeographic patterns, the impact of common historical events on phylogeographic patterns and evolutionary histories of sympatric species remains under debate (Rossetto et al. 2007).
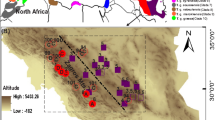
The comparative phylogeography on continental islands primarily focuses on biodiversity hotspots such as the archipelagos in the Mediterranean, North Eastern Europe, and Ryukyu of East Asia (e.g., Papadopoulou et al. 2009; Weese et al. 2013; Suzuki et al. 2014; Parmakelis et al. 2015; Kubota et al. 2017). To date, few studies have been conducted on the continental islands of China, despite the fact that tens of thousands of islands are located on the coastal continental shelf, reaching from the tropical to the temperate zone of the Northern Hemisphere. The Zhoushan Archipelago is located in the northeast of Zhejiang Province in East China Sea (Fig. 1). The archipelago consists of 1339 islands and thus constitutes the largest archipelago in China. These islands were originally part of the Tiantai Mountain range. They were separated from the mainland following the rise of sea levels during the late Pleistocene and were totally isolated about 7–9 kya during the Holocene (Wang et al. 2014). The Zhoushan Archipelago is characterized by relatively low species richness and a small geological setting. To date, few species have been investigated on the islands adjacent to the mainland (Hu et al. 2006; Chen et al. 2008). Recently, Wang et al. (2014) studied the influence of geographic isolation on the genetic diversity between the mainland and island populations of pond frogs (Pelophylax nigromaculatus) across the entire Zhoushan Archipelago. The authors hypothesized that the decreased genetic diversity and genetic differences among populations may have been caused by the geographic isolation of islands. However, the impact of geographic isolation on the phylogeographic pattern of sympatric species in the Zhoushan Archipelago has not been evaluated. A growing body of archaeological evidence (e.g., woody paddles and ceramic boat) indicates that, in addition to natural causes, humans were active in the area for at least 5400 years (Zhao 2007; Zheng et al. 2016). Human activities can affect the genetic structure and population demography of species. For example, phylogeographic analysis of the weasel (Mustela nivalis) in the Western-Palearctic region indicated that human intervention resulted in population dispersal across the Mediterranean islands (Lebarbenchon et al. 2010). Similarly, species colonization mediated by human activities was also reported for the Oriental fire-bellied toad (Bombina orientalis) in Northeast Asia (Fong et al. 2016). This study suggested that rice cultivation may have facilitated the expansion of the toad population by providing additional breeding habitat for the species. Therefore, the Zhoushan Archipelago provides a suitable model ecosystem to investigate the combined influence of geographic isolation and human activities on the evolutionary processes of sympatric species.

Sample collection of the two species and the geographical distribution of different lineages of G. distinctissima. Locations of sampling are shown as different geometric shapes. Proportion of different lineages in one population is marked in different colors based on the number of haplotypes belonging to each lineage
Geisha distinctissima (Fulgoromorpha: Flatidae) and Megacopta cribraria (Pentatomomorpha: Plataspidae) are two hemipteran insects. Both species are widely distributed in Southern China (Suiter et al. 2010; Zhang et al. 2010). Geisha distinctissima is characterized by univoltine per year, with nymphs reaching adulthood between July and October, and eggs produced during late summer/autumn for wintering (He et al. 2013). Being a polyphagous insect, this species inhabits fruit trees (e.g., orange and pear), tea plants, shrubs (e.g., Pyracantha fortuneana and Neoltisea sericea), and macrophanerophytes (e.g., Cinnamonum campora, Sapium sebiferum, and Magnolia grandiflora) (Zhang et al. 2010; Yu and Chen 2013). Many of these plants provide material for human livelihood (e.g., food, making tools, and building house), which suggests that the insect is related to human activities. M. cribraria is multivoltine with three generations per year in Southern China (Wu et al. 2006). Adults emerge from March to October and overwinter by hiding in shelters, such as cavities under the tree bark and leaf litter adjacent to their host plants. This insect is an agricultural pest and mainly damages soybean (Glycine max) and kudzu (Pueraria montana) (Suiter et al. 2010; Zhang et al. 2012). During our sample collection in the Zhoushan Archipelago, we found that the two species are sympatric in most sampling locations. Therefore, we conducted a comparative phylogeographic analysis of co-occurring G. distinctissima and M. cribraria based on mitochondrial (mtDNA) and nuclear DNA (nDNA) to investigate the impact of geographic isolation and human activities on their evolutionary histories in the Zhoushan Archipelago.
Materials and methods
Taxon sampling and DNA sequencing
In total, 496 specimens (G. distinctissima: 247; M. cribraria: 249) were collected from 17 locations in the Zhoushan Archipelago (14 locations of 12 islands representing the entire range of the archipelago) and adjacent mainland (three locations) (Table 1 and Fig. 1). All samples were preserved in 95% ethyl alcohol and stored at −20 °C. Genomic DNA was extracted from leg tissues using a Blood & Tissue Geniomic DNA Kit (TIANGEN, Beijing). The mitochondrial cytochrome oxidase subunit 1 (COX1), cytochrome c oxidase subunit 2 (COX2) to cytochrome c oxidase subunit 3 (COX3), including two tRNA (Lys and Asp) and two ATP synthase F0 subunits (ATP6 and ATP8), and cytochrome b (CYTB) were amplified using specific primers (Table S1 in Appendix S1). PCR was performed in 25 μL volume containing 12.5 μL Premix Taq™ (Takara Bio, Dalian), 0.8 μL of each primer, 0.6 μL DNA template, and 10.3 μL ddH2O. The thermal regime consists of 5 min initial denaturation at 94 °C; followed by 35 cycles of 30 s denaturation at 94 °C, 40 s annealing at 50–55 °C, and 1 min extension at 72 °C; and a final extension for 10 min at 72 °C. Nuclear fragment ITS2 and the D2 variable region of 28S rDNA were also amplified following the same reaction protocols with annealing at 55–60 °C. PCR products were examined by 1% agarose gel electrophoresis, and suitable products were sequenced using an ABI 3730 automated sequencer (TsingKe, Beijing). Several PCR products with multiple bands were purified and cloned with pDM19-T as vector and Trans5α cell as competent cell following standard protocols. Four clones of plasmid DNA of each sample were sequenced by universal M13 primers. DNA sequences were assembled by SeqMan in DNAStar Lasergene v7.1.0 package (DNASTAR, Madison, WI, USA) and aligned by MUSCLE in MEGA 7 (Kumar et al. 2016). In order to avoid false nucleotide polymorphism, several sites with missing data in sequences were discarded. All newly obtained sequences have been submitted to GenBank with accession numbers MK427708-MK429678 (Tables S2 and S3 in Appendix S1).
Genetic diversity and population genetic structure
The haplotype and genetic diversity of each sampling location [including the number of haplotype (Nh), haplotype diversity (Hd), and nucleotide diversity (π)] were calculated using DNASP v5.10.01 (Librado and Rozas 2009). Several approaches were independently employed to estimate the population genetic structure of the two species. A Bayesian inference method was firstly employed using BAPS v6.0 software (Cheng et al. 2013). In order to ensure stabilized clustering, the model for spatial clustering of groups with 20 runs was used. Median-joining network analysis of haplotypes with default parameters was conducted to estimate the population genetic structure using Network v4.6 (Bandelt et al. 2000). In addition, hierarchical analysis of molecular variance (AMOVA) was used to further test the validity of the predefined lineages in Arlequin v3.5 (Excoffier and Lischer 2010).
Phylogeographic patterns and estimation of divergence time
The software package SplitsTree v4 (Huson and Bryant 2005) provides a framework for evolutionary analysis using both trees and networks based on P-distance. It was therefore employed to identify phylogenetic signals of both mtDNA and nDNA haplotypes. Next, the phylogenetic tree among populations of the two species was reconstituted using MrBayes v3.2.6 (Ronquist et al. 2011) through the online CIPRES workspace (http://www.phylo.org). Recently, Hosokawa et al. (2014) used a species of Coptosoma as the outgroup and successfully determined the origin of invading M. cribraria in North America. Therefore, C. bifaria (GenBank accession: EU427334) was selected as the outgroup for M. cribraria populations. In addition, phylogenetic analysis of the family Flatidae based on mtDNA showed that Salurnis marginellus was located in a clade more basal than that of G. distinctissima, with relatively high branch support values (unpublished data). Thus, S. marginellus was selected as the outgroup for G. distinctissima populations. In order to avoid overparameterization leading to bias estimation of bipartition posterior probabilities (Lemmon and Moriarty 2004) or nonidentifiability of model parameters (Marshall et al. 2006), the combined mtDNA and combined nDNA were independently employed for phylogenetic analysis. The best-fit models of nucleotide substitution were selected by the jModelTest v2.1.7 (Darriba et al. 2012) based on Akaike information criterion, Bayesian information criterion, and decision theoretic performance-based approach. The optimal models, determined by at least two model-selecting approaches, were implemented for phylogenetic analysis. For MrBayes analysis, two independent runs were conducted for 100 million generations with trees sampled for every 10,000 generations. The first 25% of the sampled trees were discarded as burn-in. The effective sampling sizes (ESSs > 200) and the posterior distribution were examined by Tracer v1.6 (Rambaut et al. 2014). The maximum clade credibility tree with posterior clade probability was obtained with TreeAnnotator v1.8.4 in the BEAST package (Drummond et al. 2012). The divergence time was estimated using a coalescence-based model with an uncorrelated lognormal relaxed clock and constant size tree prior in BEAST v1.8.4 (Drummond et al. 2012). The relaxed clock model allows different clock rates among tree branches. The analysis was performed using the proposed conventional mutation rate of insect COX1 gene of 2.3% per million years (Brower 1994). The analysis on both species ran 10 million generations, with trees sampled for every 10,000 generations and the first 10% of samples as burn-in. The ESSs and the posterior distribution were monitored by Tracer v1.6. The tree and divergence time were visualized using FigTree v1.3.1.
Ancestral range assessment
In order to evaluate the ancestral range of the two species, the distribution of both species was subdivided into four geographical areas based on the distance of the islands from the mainland or Zhoushan Island (Table 1). S-DIVA and Bayesian Binary MCMC (BBM) methods were used to estimate the ancestral distribution using the RASP (Reconstruct Ancestral State in Phylogenies) software (RASP v3.0; Yu et al. 2015). S-DIVA is a modified version of the Dispersal–Vicariance Analysis (DIVA) model, which can calculate the occurrence of an ancestral range at a node using the frequency of all alternative reconstructions (Yu et al. 2010). BBM is primarily designed for the reconstruction of ancestral range, with the probabilities of each unit area in a given node generated by MrBayes. For our purpose, the maximum clade credibility tree obtained by phylogenetic reconstruction was employed for both methods with default parameters, and the setting of Jukes-Cantor model + G in BBM was as per a previous study (Perrot‐Minnot et al. 2018).
Population demography and gene flow
Neutrality analysis was conducted to investigate the demographic history of the two species. Under the assumption of neutrality, the expanded population usually exhibits negative values for both Tajima’s D and Fu’s FS. Pairwise mismatch distributions were also conducted to confirm the historical demography of the two species. It has previously been shown that the unimodal mismatch distribution represents population expansion, while multimodal formats represent populations whose size remains constant (Rogers and Harpending 1992). In addition, statistical parameter raggedness (Rg) index and the sum of square deviations (SSD) were also calculated to further confirm the demographic history of both species. A small value of the Rg index indicates an expansion event of populations, and a significant SSD value shows the stability of the population size (Dsouli-Aymes et al. 2011). Both indices were calculated in Arlequin v3.5. The population size dynamics over time was estimated with the best-fit models of nucleotide substitution using Bayesian Skyline Plot (BSP) in BEAST. The Markov Chain was used to run 100 million generations, with trees sampled for every 10,000 steps and the first 25% samples as burn-in. The coalescent Bayesian skyline method was conducted under an uncorrelated lognormal relaxed clock model. The parameter estimates and ESS values were monitored by Tracer v1.6.
The gene flow among the defined lineages of the two species was estimated. The mutation-scaled population size (θ; θ = Neμ, where Ne is theeffective population size and μ themutation rate per generation) and the mutation-scaled migration rate (M; M = m/μ, where m is themigration rate) were calculated using Bayesian inference in Migrate v3.6 (Beerli and Felsenstein 2001). Parameters were set as long-inc = 20, long-steps = 1,000,000, burn-in = 100,000. To increase the efficiency of the MCMC, four heating chains from cold to boiling were used with parameter setting at 1, 1.5, 3, and 10,000. Although the Bayesian inference is more efficient in searching parameter space than the ML approach, the convergence among runs remains uncertain (Beerli 2005). Therefore, each run was repeated three times. The mean values of the average M were counted, and their statistical differences were examined by T-tests to evaluate dispersal patterns among defined lineages for the two species.
Results
Genetic diversity and population genetic structure
We obtained a 2811/2604 bp combined mtDNA fragment (COX1: 840/793 bp, COX2-COX3: 1110/1062 bp, and CYTB: 861/749 bp) without pseudogenes and nuclear copies from 242/249 individuals in G. distinctissima and M. cribraria, respectively. A 650-bp combined nDNA fragment (28S rDNA:125 bp, ITS2: 525 bp) was obtained from G. distinctissima (sequences for 28S rDNA are listed in Appendix S2). For unknown reasons, we failed to obtain the ITS2 fragment in M. cribraria even after multiple attempts. By following our protocol, we only obtained a 488-bp fragment of 28S rDNA from all sampled individuals in M. cribraria. Eighty-four (in G. distinctissima) and 127 (in M. cribraria) haplotypes were identified using the combined mtDNA fragment in the two species (Tables S4 and S5 in Appendix S1). In contrast, relatively few haplotypes (25 in G. distinctissima and 4 in M. cribraria) were obtained in the two species when using nDNA fagment (Tables S6 and S7 in Appendix S1), which suggests that the nDNA fragment is highly conserved in both species.
BAPS analysis based on mtDNA showed that three distinct lineages (L1–L3) were recognized in G. distinctissima (Fig. 2a). However, these three lineages did not exhibit clear geographical patterns. Populations from MAI (mainland) distributed into L2 (XY) and L3 (RYS), and clustered with populations from AMI (adjacent-mainland islands: LHJT, LHTM, TH, JT, ZW, QTS, ZJJ). Populations from AMI scattered across all three lineages without clear geographical separation. Interestingly, populations from AZI (islands adjacent to Zhoushan Island: XS, DS, DJ, QS) and RIS (remote islands: GQ & SJ) were isolated from each other. The former nested into L2, while the latter nested into L1. Consistent with our BAPS analysis, the mtDNA haplotype network also identified the three lineages (L1–L3) divided by several missing haplotypes and mutations (Fig. 2b). Most haplotypes in L1 and L2 were derived from the ancestral haplotype H_1 and H_15 in one or two steps. Geographical mapping of the three lineages showed that they also lacked observable geographical separation (Fig. 1). In addition, we identified two lineages in G. distinctissima by BAPS analysis using the nDNA fragment (Fig. S1 in Appendix S3). Nevertheless, the haplotype network derived by nDNA fragment displayed a star-shaped network without any discernable genetic structure (Fig. 2c), which suggests that the division of the two lineages in G. distinctissima is unreliable for the conservation of nDNA fragment. BAPS analysis based on both mtDNA and nDNA failed to identify any distinguishable genetic structure among the populations of M. cribraria (data not shown). Additional network analysis using mtDNA haplotypes showed that populations of M. cribraria presented only one lineage with a star-shaped network (Fig. 3a). Most haplotypes were derived from a few ancestral haplotypes (H_5, H_8, H_18, H_29, H_34 and H_47), which suggests potential recent expansion events of M. cribraria. Network analysis on nDNA of M. cribraria showed that haplotype H_1 was shared by almost all individuals with only three derived haplotypes (Fig. 3b).

Genetic structures of G. distinctissima based on mtDNA and nDNA. a BAPS analysis based on mtDNA; b network map based on mtDNA; c network map based on nDNA. The size of circles represents the number of individuals for the haplotype. The numbers in the square brackets are mutations

Network map of M. cribraria. a network map based on mtDNA; b network map based on nDNA. The circle size represents the number of individuals for the haplotype
For mtDNA, AMOVA analysis of the three lineages in G. distinctissima showed that the largest variation was distributed among lineages (74.1% P < 0.001) (Table 2), which suggests that the three lineages exhibit strong genetic divergence. When comparing only AZI and RIS groups, we found that they exhibit significant genetic difference, with ~50% variation (P < 0.001) (Table 2). In contrast, the largest variation (95.55% variation, P < 0.001) of nDNA occurs within G. distinctissima populations (Table 2). However, the variations of both mtDNA and nDNA were distributed within M. cribraria populations (Table 2), indicating that M. cribraria exhibits little genetic divergences among populations. In addition, it seems that the genetic diversity among MAI, AMI, AZI, and RIS for both species differs on the basis of mtDNA (Table 1). However, AMOVA analysis for the four groups showed the largest genetic variation for both species distributed within groups (Table S8), which suggests that populations among the four regions exhibit little genetic difference.
Phylogeographic patterns, divergence time, and ancestral distribution
Our SplitsTree analysis for G. distinctissima also identified three distinct lineages on the basis of mtDNA haplotypes (Fig. S2 in Appendix S3). The monophyly of the three lineages was further verified by phylogenetic analysis based on mtDNA haplotypes (the best-fit model: HKY + I + G) (Fig. 4). However, we failed to determine the exact relationship among the three lineages due to their relatively low support values. Our haplotype network of the mtDNA fragment shows that L3 is divergent from the other two lineages, while the phylogenetic tree of the three lineages shows that L1 is the basal clade. The divergence time showed that the three lineages began to separate at around 13.7 kya (95% HPD: 13.5–13.8 kya) and thoroughly formed before 8.9 kya (95% HPD: 7.4–10.4 kya). S-DIVA analysis showed that the potential ancestral range of G. distinctissima lay in the AMI + RIS region with an occurrence frequency of 100% (Fig. S3a in Appendix S3). Although the BBM method performed poorly (Fig. S3b in Appendix S3), this result suggests that the ancestral range of G. distinctissima may originate in the AMI + RIS region. Similar to the results of the other two analyses (BAPS and network analysis) derived from nDNA, SplitsTree analysis based on the same fragment also failed to identify observable genetic lineages in G. distinctissima (Fig. S4 in Appendix S3). Therefore, we omitted the phylogenetic analysis, analysis of divergence time, and ancestral range evaluation using the nDNA fragment.

The phylogenetic tree and divergence times of G. distinctissima based on mtDNA. The numbers on the top of the bar show the posterior probability and the numbers below represent the divergence time with 95% HPD
Both SplitsTree and phlylogenetic analysis based on mtDNA haplotypes (the best-fit model: HKY + I + G) showed that populations of M. cribraria failed to yield discernible genetic structures (Figs. S5 and S6 in Appendix S3). As the phylogenetic signal of the mtDNA fragment was insufficient, we also omitted the estimation of divergence time among populations of M. cribraria. Analysis of ancestral range showed that both methods (S-DIVA and BBM) performed poorly in determining the ancestral distribution of M. cribraria (Fig. S7 in Appendix S3). Nevertheless, these results suggest that the potential ancestral range lies in the MAI or MAI + AMI region. In addition, only four haplotypes were identified on the basis of nDNA in M. cribraria, with most individuals sharing haplotype H_1. This observation suggests that the amount of variation based on nDNA is insufficient to estimate the genetic structure and divergence time.
Demographic history and gene flow
Neutrality tests on both species identified significant negative values of Tajima’s D and Fu’s FS (except nDNA in M. cribraria) with P < 0.05 (Table S9 in Appendix S1), which suggests that the two species may have undergone population expansion. Although the multimodal format of mismatch distribution was identified based on mtDNA in G. distinctissima, the small Rg index and the lack of significant SSD imply that populations of G. distinctissima exhibit recent expansion events (Fig. 5a). The BSP analysis showed that the size of G. distinctissima populations increased rapidly before the genetic divergence of the three lineages (L1–L3), and a mild expansion event occurred at 5–9 kya (Fig. 5b). In M. cribraria, a canonical unimodal mismatch distribution with a small Rg index and no significant SSD was identified using mtDNA, which indicates that the species had undergone a more recent population expansion (Fig. 5c). The BSP analysis also shows that M. cribraria experienced a slow increase in population size until ~7.5 kya, and since then maintained a stable population size (Fig. 5d).

Mismatch distribution and Bayesian skyline plots (BSP) analysis of the two species based on mtDNA. a and c represent the mismatch distribution for G. distinctissima and M. cribraria, respectively; b and d represent the Bayesian skyline plots for G. distinctissima and M. cribraria, respectively
To estimate the dispersal patterns of the two species in the Zhoushan Archipelago, the gene flow among different regions was analyzed using mtDNA (Fig. 6 and Table S10 in Appendix S1). As shown in Fig. 6, a markedly asymmetric gene flow occurred in G. distinctissima from AMI to MAI. For M. cribraria, however, we identified an antipodal gene flow direction. To estimate the gene flow of the entire region in the Zhoushan Archipelago and its adjacent mainland, we combined the regions of MAI and AMI. Our estimation clearly showed that both species exhibited frequent asymmetrical gene flow from MAI + AMI to distant islands. Furthermore, the migration rates distinctly increased in recent 5 kya, which suggests that the frequent asymmetrical gene flow occurred no earlier than 5 kya in both species (Fig. S8 in Appendix S3).

Migration (M) of the two insects among different regions based on mtDNA. M indicates the mutation-scaled migration rate. MAI mainland, ZIS the Zhoushan Island (including ZJJ), RTL regions including TH, LHTM, LHJT, and JT (this region includes only TH and LHTM for M. cribraria), AMI adjacent-mainland islands including islands of ZIS and RTL regions, AZI islands adjacent to Zhoushan Island, RIS remote islands
Discussion
In summary, our study clearly demonstrates that, although G. distinctissima and M. cribraria exhibit concordant historical demographies, they feature contrasting genetic structures in the Zhoushan Archipelago. During our analysis, we found that the mtDNA fragment contains more signal sites than the nDNA fragment, which provides relatively high resolution for the genetic structures and the demographic history of the two species. It has previously been reported that, due to its maternal inheritance, mtDNA alone cannot reveal the complete historical processes of a species with different evolutionary histories between males and females due to wing polymorphism (Gäde 2002). However, no reports have been published to date about the differences in wing morphology and dispersal ability between males and females of both G. distinctissima and M. cribraria. Therefore, although our hypothesis is mainly derived from the analysis of our mtDNA dataset, it clearly reflects the complete evolutionary history of the two species.
Frequent asymmetric gene flow of the two species
Our comparative analysis shows that both G. distinctissima and M. cribraria exhibit little genetic divergences among populations located in the Zhoushan Archipelago and the adjacent mainland. Mainland populations usually display higher genetic diversity than the island populations (Frankham 1997). A recent comparative analysis on pond frogs (P. nigromaculatus) in the Zhoushan Archipelago also put forward the same conclusion (Wang et al. 2014). This study suggested that the genetic differences between mainland and island populations may be shaped by island isolation due to the rise of sea level during the Holocene. However, our results indicate that both insect species underwent frequent asymmetrical gene flow events, starting from the mainland and adjacent islands region, and spreading toward more distant islands, which may have resulted in genetic homogenization of the populations on the mainland and across the Zhoushan Archipelago. Furthermore, the gene flow occurred no earlier than 5 kya, and exhibited an increasing trend over time. Previous archeological studies have suggested that humans dwelled in the Zhoushan Archipelago for at least 5400 years (Zhao 2007; Zheng et al. 2016). Subsequently, human activities between the Zhoushan Archipelago and mainland increased with the development of shipbuilding in China (Fang 2008). Therefore, human activities may have resulted in the frequent asymmetrical gene flow among populations for both species. In addition, most of the host plants for the two species are vital for the development of human civilization. Stout shrubs and arbors can be used to build houses and manufacture paddles, boats, and rafts. Despite the lack of adequate documentation for kudzu, numerous archaeological studies and historical records have shown that the utilization of kudzu and soybean by humans has a history of more than 5000 years in China (Xiao et al. 2013; Sun 2014). It also has been reported that M. cribraria usually travels long distances due to related human activities by attaching to the surface of clothing, goods, and vehicles (Takano and Takasu 2016). Together, these findings further suggest that human activities may result in the frequent asymmetrical gene flow and subsequently homogenize the gene pool between populations of the two species in the Zhoushan Archipelago and adjacent mainland.
It has been reported previously that other factors such as monsoons, ocean currents, and dispersal ability of the species facilitate gene flow among populations isolated by geographical barriers (Chiang et al. 2006; Tseng et al. 2018). The East China Sea contains ocean currents that flow from south to north (Ni et al. 2014), which may aid the dispersal of the two winged insects from mainland to the Zhoushan Archipelago via drifting. However, neither species exhibits visible gene flow between the mainland and the Zhoushan Archipelago prior to 5 kya (Fig. S8 in Appendix S3), suggesting that dispersal by flight and ocean current had negligible effect on the frequent asymmetrical gene flow before the beginning of human activities. Furthermore, the Zhoushan Archipelago is located in the subtropical zone, which experiences prevailing northern or southeast trade winds throughout the year (Ye 2000). Thus, the direction of the trade wind is inconsistent with the gene flow direction from the mainland to adjacent islands.
Contrasting genetic structures of the two species
Although our two sympatric species failed to exhibit clear phylogeographic patterns, they present contrasting genetic structures in the Zhoushan Archipelago. Multiple lines of evidence indicate that G. distinctissima formed three distinct genetic lineages (L1–L3). This phenomenon was also identified in Chinese Water Deer (Hydropotes inermis inermis), which showed significant genetic differentiation without visible geographical separation (Hu et al. 2006). According to a previous hypothesis, the genetic discontinuities with the lack of spatial separation may have arisen from secondary contact among populations (Avise et al. 1987). The ancestral distribution of G. distinctissima in the AMI + RIS regions hints that the ancestral population may have widely distributed in the Zhoushan Archipelago. The Zhoushan Archipelago was once covered by abundant arbors and shrubs (Ye 2000; Chen et al. 2010). Most of those plants (e.g., Lauraceae, Euphorbiaceae, Celastraceae, and Aquifoliaceae) are known to be hosts to G. distinctissima (Zhang et al. 2010; Yu and Chen 2013), which also suggests that their ancestral populations were once widely distributed in the area. The divergence time estimation showed that the three lineages began to split ~13.7 kya (Fig. 4) ago, and they all formed at 8.9 kya in spite of their uncertain phylogenetic relationship. This suggests that the three lineages had completely diverged at 8.9–13.7 kya. During this period, the sea level of East China Sea continuously rose from ∼−70 to −20 m, and geographically isolated several of the islands (Zhang et al. 2013). This isolation possibly blocked the gene flow and resulted in the genetic differentiation among populations of G. distinctissima. Thereafter, human-mediated secondary contact among divergent populations and the frequent asymmetrical gene flow from the mainland and adjacent islands to distant islands may have resulted in the collapse of spatial separation and the uncertain relationship between the three lineages in G. distinctissima. Thus, on the basis of these results, we hypothesize that the combined roles of island isolation and human activity in the region shaped the three lineages without clear geographical separations in G. distinctissima. Our analysis showed that, in contrast to G. distinctissima, M. cribraria exhibits only one single lineage. The location of the ancestral range in the MAI or MAI + AMI region suggests that the ancestral population of M. cribraria did not distribute in the Zhoushan Archipelago or just inhabit in the islands adjacent to the mainland. This indicates that M. cribraria colonized into the entire archipelago after it spread from the mainland or adjacent islands. Furthermore, our analysis showed that the frequent asymmetrical gene flow from the mainland and adjacent islands to distant islands occurred in <5 kya. This time period overlaps with the period of human activities in the region. Therefore, human activities may have resulted in the spread of M. cribraria from the mainland to the Zhoushan Archipelago. In addition, the persistent human activities in the mainland and other islands may have further increased gene flow and homogenized gene pools among populations in M. cribraria.
It has been verified that sympatric species with concordant phylogeographic patterns exhibit a long-standing geographical association and shared biogeographic history (Arbogast and Kenagy 2001). The distinct ancestral ranges of G. distinctissima and M. cribraria show that they are not historically sympatric, which suggests that the two insect species have different biogeographic histories and relatively short geographical association. It has been reported that most currently sympatric species share no common biogeographic histories, which also causes the phylogeographic differences (Arbogast and Kenagy 2001). Therefore, although no clear phylogeographic patterns could be deteremined for the two species, the contrasting genetic structures suggest that they responded differently to the shared historical factors (island isolation and human activities) and had independent evolutionary histories in the Zhoushan Archipelago. Numerous studies also have shown that many sympatric species exhibited different genetic structures and geographical subdivision (Hurtado et al. 2004; Michaux et al. 2005; Rossetto et al. 2007; Crandall et al. 2010). It has been hypothesized that these differences are associated with the dispersal abilities, host preferences, and biological traits of organisms. The evolution of organisms is an extremely long and complicated process that involves multiple aspects of genetic organization, ecological requirement, biological feature, biogeographic histories, and their interactions. Any biotic or abiotic element (e.g., the habitat preferences and dispersal abilities of species, climate oscillation, geological barriers, and biogeographic differences) can affect the evolutionary trajectory of organisms. Therefore, a comparative analysis of multiple regions, larger sample numbers, and multiple factors should be performed to better understand the evolutionary histories of sympatric species.
Post-LGM population expansion of the two species
Both neutrality test and mismatch analysis showed that the two species underwent population expansion events. Our analysis indicates that a sharp increase of G. distinctissima populations occurred before the genetic divergence of the three lineages post LGM. Subsequently, the population size exhibited a mild increase at 5–9 kya. By contrast, the population of M. cribraria expanded during the post-LGM period until 7.5 kya. These expansion events occurred earlier than the event of gene flow (started no earlier than 5 kya) and human activities (from about 5.4 kya) in the Zhoushan Archipelago, which indicates that human activities and gene flow are unlikely to have caused population expansion events in both species. Previous reports have shown that during the LGM period thermophilic species were restricted in small refugia, while they stretched their ecological niches as the temperature rose during the post-LGM period (Runck and Cook 2005). In spite of these climatic fluctuations, the temperature in Eastern China continued to rise post LGM (Yang and Xie 1984), which suggests that the area is suitable for thermophilic plants. In addition, climatic fluctuations caused the sea level of the East China Sea to rise slowly and reach current levels until ~5.5 kya (Yang and Xie 1984). The relatively low sea levels may have exposed the abundant land area of continental islands and adjacent mainland to thermophilic plant colonization. It has been reported that most host plants (such as kudzu, soybean, and several species of family Lauraceae, Euphorbiaceae, Celastraceae, and Aquifoliaceae) of the two species are thermophilous (Wu and Chen 1994; Wang et al. 2009; Wang and Ye 2017). Therefore, the climatic fluctuations of Eastern China during the at post-LGM period may have provided more ecological niches for host plants and also facilitated population expansion of G. distinctissima and M. cribraria.
Conclusions
In the present study, we conducted a comparative phylogeographic analysis of two sympatric hemipteran species (G. distinctissima and M. cribraria) in the Zhoushan Archipelago and its adjacent landmass. Our results clearly demonstrate that G. distinctissima formed three distinct genetic lineages (L1–L3) with no visible geographical separation. This phylogeographical pattern is caused by the combined roles of geographical isolation of islands and the recent frequent asymmetric gene flow (from mainland and adjacent islands to other distant islands) caused by human activities. However, M. cribraria only forms one genetic lineage and exhibit no geographical separation attributable to human-mediated recent colonization from the adjacent mainland region. The contrasting genetic structures suggests that the two currently sympatric species with different biogeographic histories showed different responses to island isolation and human activities, and independent evolutionary histories in the Zhoushan Archipelago. Our demographic analysis of population histories for the two species showed that they experienced population expansion during the post-LGM period, which reflects the similar impacts of the climatic fluctuations in Eastern China on the population size of the two species. Our results reveal that the two hemipteran insects studied here exhibit contrasting genetic structures while displaying concordant historical demographies because of the shared historical factors in the Zhoushan Archipelago. Further studies with additional samples and a comprehensive consideration of multiple factors should provide a better understanding of the evolutionary patterns of the sympatric species in the Zhoushan Archipelago.
DATA ARCHIVING
Molecular sequences obtained in the present study have been deposited at NCBI (https://www.ncbi.nlm.nih.gov/nucleotide/) under the following Accession IDs MK427708-MK429678.
References
Arbogast BS, Kenagy GJ (2001) Comparative phylogeography as an integrative approach to historical biogeography. J Biogeogr 28:819–825
Avise JC, Arnold J, Ball RM, Bermingham E, Lamb T, Neigell JE, Reebl CA, Saunders NC (1987) Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annu Rev Ecol Syst 18:489–522
Bandelt HJ, Macaulay V, Richards M (2000) Median networks: speedy construction and greedy reduction, one simulation, and two case studies from human mtDNA. Mol Phylogenetics Evol 16:8–28
Beerli P, Felsenstein J (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci 98: 4563–4568
Beerli P (2005) Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 22:341–345
Bittkau C, Comes HP (2005) Evolutionary processes in a continental island system: molecular phylogeography of the Aegean Nigella arvensis alliance (Ranunculaceae) inferred from chloroplast DNA. Mol Ecol 14:4065–4083
Brower AV (1994) Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc Natl Acad Sci USA 91:6491–6495
Byun SA, Koop BF, Reimchen TE (1997) North American black bear mtDNA phylogeography: implications for morphology and the Haida Gwaii glacial refugium controversy. Evolution 51:1647–1653
Campagna L, St Clair J, Lougheed SC, Woods RW, Imberti S, Tubaro PL (2012) Divergence between passerine populations from the Malvinas–Falkland Islands and their continental counterparts: a comparative phylogeographical study. Biol J Linn Soc 106:865–879
Chen B, Wang G, Wang M, Qiu HS (2010) The wild germplasm resources of ground-covering plants in Zhoushan Archipelago. Chin Wild Plant Resour 29:47–49
Chen Y, Shi MM, Ai B, Gu JM, Chen XY (2008) Genetic variation in island and mainland populations of Ficus pumila (Moraceae) in eastern Zhejiang of China. Symbiosis 45:37–44
Cheng L, Connor TR, Sirén J, Aanensen DM, Corander J (2013) Hierarchical and spatially explicit clustering of DNA sequences with BAPS software. Mol Biol Evol 30:1224–1228
Chiang YC, Hung KH, Schaal BA, Ge XJ, Hsu TW, Chiang TY (2006) Contrasting phylogeographical patterns between mainland and island taxa of the Pinus luchuensis complex. Mol Ecol 15:765–779
Cowie RH, Holland BS (2006) Dispersal is fundamental to biogeography and the evolution of biodiversity on oceanic islands. J Biogeogr 33:193–198
Crandall ED, Frey MA, Grosberg RK, Barber PH (2010) Contrasting demographic history and phylogeographical patterns in two indo-pacific gastropods. Mol Ecol 17:611–626
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
Ditchfield AD (2000) The comparative phylogeography of Neotropical mammals: patterns of intraspecific mitochondrial DNA variation among bats contrasted to nonvolant small mammals. Mol Ecol 9:1307–1318
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973
Dsouli-Aymes N, Michaux J, De Stordeur E, Couloux A, Veuille M, Duvallet G (2011) Global population structure of the stable fly (Stomoxys calcitrans) inferred by mitochondrial and nuclear sequence data. Infect Genet Evol 11:334–342
Duryea MC, Zamudio KR, Brasileiro CA (2015) Vicariance and marine migration in continental island populations of a frog endemic to the Atlantic Coastal forest. Heredity 115:225–234
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fang M (2008) On Zhoushan’s history and culture. J Zhejiang Ocean Univ 25:60–66
Fong JJ, Li PP, Yang BT, Zhou ZY, Leaché AD, Min MS, Waldman B (2016) Influence of geology and human activity on the genetic structure and demography of the Oriental fire-bellied toad (Bombina orientalis). Mol Phylogenetics Evol 97:69–75
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity 78:311–327
Gäde G (2002) Sexual dimorphism in the pyrgomorphid grasshopper Phymateus morbillosus: from wing morphometry and flight behavior to flight physiology and endocrinology. Physiol Entomol 27:51–57
Hosokawa T, Nikoh N, Fukatsu T (2014) Fine-scale geographical origin of an insect pest invading North America. PLoS ONE 9:e89107
Hu J, Fang SG, Wan QH (2006) Genetic diversity of Chinese water deer (Hydropotes inermis inermis): implications for conservation. Biochem Genet 44:156–167
Huang JP, Lin CP (2011) Lineage-specific late pleistocene expansion of an endemic subtropical gossamer-wing damselfly, Euphaea formosa, in Taiwan. BMC Evolut Biol 11:94
Hurtado LA, Erez T, Castrezana S, Markow TA (2004) Contrasting population genetic patterns and evolutionary histories among sympatric sonoran desert cactophilic Drosophila. Mol Ecol 13:1365–1375
Huson DH, Bryant D (2005) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
He J, Yu WT, Chen W (2013) Occurrence regularity and control of Geisha distinctissima in guiyang tea plantation. Guizhou Agric Sci 41:105–107
Kubota Y, Kusumoto B, Shiono T, Tanaka T (2017) Phylogenetic properties of Tertiary relict flora in the East Asian continental islands: imprint of climatic niche conservatism and in situ diversification. Ecography 40:436–447
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lebarbenchon C, Poitevin F, Arnal V, Montgelard C (2010) Phylogeography of the weasel (Mustela nivalis) in the Western-Palaearctic region: combined effects of glacial events and human movements. Heredity 105:449–462
Lejeusne C, Bock DG, Therriault TW, MacIsaac HJ, Cristescu ME (2011) Comparative phylogeography of two colonial ascidians reveals contrasting invasion histories in North America. Biol Invasions 13:635–650
Lemmon AR, Moriarty EC (2004) The importance of proper model assumption in Bayesian phylogenetics. Syst Biol 53:265–277
Librado P, Rozas J (2009) DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Luo D, Yue JP, Sun WG, Xu B, Li ZM, Comes HP, Sun H (2016) Evolutionary history of the subnival flora of the Himalaya‐Hengduan Mountains: first insights from comparative phylogeography of four perennial herbs. J Biogeogr 43:31–43
Lyons SK (2003) Quantitative assessment of the range shifts of Pleistocene mammals. J Mammal 84:385–402
Marshall DC, Simon C, Buckley TR (2006) Accurate branch length estimation in partitioned Bayesian analyses requires accommodation of among-partition rate variation and attention to branch length priors. Syst Biol 55:993–1003
Michaux JR, Libois R, Filippucci MG (2005) So close and so different: comparative phylogeography of two small mammal species, the yellow-necked fieldmouse (Apodemus flavicollis) and the woodmouse (Apodemus sylvaticus) in the Western-Palearctic region. Heredity 94:52–63
Ni G, Li QI, Kong L, Yu H (2014) Comparative phylogeography in marginal seas of the northwestern Pacific. Mol Ecol 23:534–548
Papadopoulou A, Anastasiou I, Keskin B, Vogler AP (2009) Comparative phylogeography of tenebrionid beetles in the Aegean Archipelago: the effect of dispersal ability and habitat preference. Mol Ecol 18:2503–2517
Parmakelis A, Rigal F, Mourikis T, Balanika K, Terzopoulou S, Rego C, Amorim IR, Crespo L, Pereira F, Triantis KA, Whittaker RJ, Whittaker RJ (2015) Comparative phylogeography of endemic Azorean arthropods. BMC Evolut Biol 15:250
Paulay G (1994) Biodiversity on oceanic islands: its origin and extinction. Am Zool 34:134–144
Perrot‐Minnot MJ, Špakulová M, Wattier R, Kotlík P, Düşen S, Aydoğdu A, Tougard C (2018) Contrasting phylogeography of two Western Palaearctic fish parasites despite similar life cycles. J Biogeogr 45:101–115
Rambaut A, Suchard M, Xie W, Drummond A (2014) Tracer v. 1.6. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, Scotland
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Ronquist F, Huelsenbeck J, Teslenko M (2011) Draft MrBayes version 3.2 manual: tutorials and model summaries. http://brahms.biology.rochester.edu/software.Html.
Rossetto M, Crayn D, Ford A, Ridgeway P, Rymer P (2007) The comparative study of range-wide genetic structure across related, co-distributed rainforest trees reveals contrasting evolutionary histories. Aust J Bot 55:416–424
Runck AM, Cook JA (2005) Postglacial expansion of the southern red-backed vole (Clethrionomys gapperi) in North America. Mol Ecol 14:1445–1456
Suiter DR, Eger JE, Gardner WA, Kemerait RC, All JN, Roberts PM, Greene JK, Ames LM, Buntin GD, Douce GK (2010) Discovery and distribution of Megacopta cribraria (Hemiptera: Heteroptera: Plataspidae) in northeast Georgia. J Integr Pest Manag 1:1–4
Sun YG (2014) From historical documents to archaeological data: origin of cultivated soybean. Soybean Sci 33:124–127
Suzuki T, Kitano T, Tojo K (2014) Contrasting genetic structure of closely related giant water bugs: phylogeography of Appasus japonicus and Appasus major (Insecta: Heteroptera, Belostomatidae). Mol Phylogenetics Evol 72:7–16
Taberlet P, Fumagalli L, Wust-saucy AG, Cosso JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Takano SI, Takasu K (2016) Ability of Megacopta cribraria (Hemiptera: Plataspidae) to cling to different surfaces against extreme wind. J Insect Behav 29:256–261
Tseng HY, Huang WS, Jeng ML, Villanueva RJT, Nuñeza OM, Lin CP (2018) Complex inter‐island colonization and peripatric founder speciation promote diversification of flightless Pachyrhynchus weevils in the Taiwan-Luzon volcanic belt. J Biogeogr 45:89–100
Tsukada M (1988) Japan: glacial and holocene vegetation history-20 ky to present, 3rd edn. Kluwer, London, p 459–518
Wang GM, Ye B (2017) Floristic composition and diversity of typical plant community in Zhoushan Archipelago, East China. Chinese. J Ecol 36:349–358
Wang GM, Zhao CL, Chen YP, Cheng C, Zhao Y (2009) National key protected plant flora and distribution characteristic in Zhoushan Islands. J Zhejiang For Sci Technol 29:43–47
Wang S, Zhu W, Gao X, Li X, Yan S, Liu X, Yang J, Gao ZX, Li YM (2014) Population size and time since island isolation determine genetic diversity loss in insular frog populations. Mol Ecol 23:637–648
Weese DA, Fujita Y, Santos SR (2013) Multiple colonizations lead to cryptic biodiversity in an island ecosystem: comparative phylogeography of anchialine shrimp species in the Ryukyu Archipelago, Japan. Biol Bull 225:24–41
Weigelt P, Kreft H (2013) Quantifying island isolation-insights from global patterns of insular plant species richness. Ecography 36:417–429
Wu DJ, Chen ZY (1994) Research on Pueraria DC. J Trop Subtrop Bot 3:12–21
Wu MX, Wu ZQ, Hua SM (2006) A Preliminary study on some biological characters of globular stink bug, Megacopta cribraria and its two egg parasitoids[J]. J Fujian Agric For Univ 35:147–150
Xiao SX, Li AP, Fan SC, Sun B (2013) Research progress of germplasm resources of Radix pueratiae. J Shanxi Agric Sci 41:99–102
Yang Y, Li J, Yang S, Li X, Fang L, Zhong C, Duke NC, Zhou RC, Shi S (2017) Effects of Pleistocene sea-level fluctuations on mangrove population dynamics: a lesson from Sonneratia alba. BMC Evolut Biol 17:22
Yang H, Xie Z (1984) Sea-level changes along the east coast of china over the last 20,000 years. Oceanol et Limnol Sin 15:1–13
Ye XY (2000) The anthophyta assemblages and paleoclimate variation of Zhoushan Island since middle Pleistocene. Mar Geol Quat Geol 20:9–13
Yu W, Chen W (2013) Study on the behavior of Geisha distinctissima Walker. J Mt Agric Biol 32:123–127
Yu Y, Harris AJ, He X (2010) S-DIVA (statistical dispersal-vicariance analysis): a tool for inferring biogeographic histories. Mol Phylogenetics Evol 56:848–850
Yu Y, Harris AJ, Blair C, He X (2015) RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol Phylogenetics Evol 87:46–49
Zhang HS, Chen Q, Sun JS (2013) Seafloor landforms in the East China Sea. Shanghai Land Resour 34:46–52
Zhang Y, Hanula JL, Horn S (2012) The biology and preliminary host range of Megacopta cribraria (Heteroptera: Plataspidae) and its impact on kudzu growth. Environ Entomol 41:40–50
Zhang Y, Peng LF, Wang YL (2010) Review of the planthopper genus Amasha Medler (Hemiptera: Fulgoromorpha: Flatidae: Phyllyphantini) with description of one new species from China. Zootaxa 2664:61–68
Zhao LP (2007) A brief Survey on the evolution and development of Zhoushan’s marine culture. J Zhejiang Ocean Univ 24:30–37
Zheng LB, Hao XD, Zhuo B, Liu YL, Wang XL, Zhu LX, Yu XB, Zhang YJ (2016) Holocene palaeoenvironment evolution and human activity of the Hemudu-Tianluoshan Sites in Yuyao of Zhejiang Province. J Palaeogeogr 18:779–983
Acknowledgements
We gratefully thank Prof. Yi-Ming Li (Institute of Zoology, Chinese Academy of Sciences) for valuable suggestions for the design of our subject. We thank Jing-Yu Liang (Institute of Entomology, College of Life Sciences, Nankai University) for sample collection and the guidance of data analysis. We also thank Fang Yu (Institute of Zoology, Chinese Academy of Sciences) for assistance with laboratory experiments. The work was supported by the National Natural Science Foundation of China (Grant nos. 31572298, 31872279).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, K., Lin, CP. & Liang, AP. Comparative phylogeography of two hemipteran species (Geisha distinctissima and Megacopta cribraria) in the Zhoushan Archipelago of China reveals contrasting genetic structures despite concordant historical demographies. Heredity 124, 207–222 (2020). https://doi.org/10.1038/s41437-019-0265-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0265-4
