
Abstract
In wide-ranging taxa with historically dynamic ranges, past allopatric isolation and range expansion can both influence the current structure of genetic diversity. Considering alternate historical scenarios involving expansion from either a single refugium or from multiple refugia can be useful in differentiating the effects of isolation and expansion. Here, we examined patterns of genetic variability in the trans-continentally distributed painted turtle (Chrysemys picta). We utilized an existing phylogeographic dataset for the mitochondrial control region and generated additional data from nine populations for the mitochondrial control region (n = 302) and for eleven nuclear microsatellite loci (n = 247). We created a present-day ecological niche model (ENM) for C. picta and hindcast this model to three reconstructions of historical climate to define three potential scenarios with one, two, or three refugia. Finally, we employed spatially-explicit coalescent simulations and an approximate Bayesian computation (ABC) framework to test which scenario best fit the observed genetic data. Simulations indicated that phylogeographic and multilocus population-level sampling both could differentiate among refugial scenarios, although inferences made using mitochondrial data were less accurate when a longer coalescence time was assumed. Furthermore, all empirical genetic datasets were most consistent with expansion from a single refugium based on ABC. Our results indicate a stronger role for post-glacial range expansion, rather than isolation in allopatric refugia followed by range expansion, in structuring diversity in this species. To distinguish among complex historical scenarios, we recommend explicitly modeling the effects of range expansion and evaluating alternate refugial scenarios for wide-ranging taxa.
Similar content being viewed by others


Introduction
Shifts in climate during the late Quaternary altered biogeographic patterns of species diversity and endemism (Sandel et al. 2011), and traces of these historical processes are embedded in current patterns of genetic diversity (Hewitt 2000). Phylogeographic analysis therefore represents a promising tool for investigating mechanisms by which climatic shifts shape biodiversity (Riddle 2016). Genetic profiles of many Nearctic and Palearctic taxa, for example, were strongly impacted by isolation in refugia during glacial episodes (Hewitt 2000). The existence of deeply divergent, spatially restricted clades within continuously distributed taxa has often been interpreted as a signal of glacial isolation in multiple allopatric refugia followed by post-glacial range expansion (Provan and Bennett 2008).
Even so, fully understanding the pattern and process of Quaternary range shifts using genetic data can be challenging, as multiple historical factors can contribute to current genetic variability. Simulations have demonstrated that range expansion, for example, can generate levels of genetic differentiation within the recolonized range that are equal to or greater than expected in cases of secondary contact between reciprocally isolated populations (Knowles and Alvarado-Serrano 2010), complicating the interpretation of genetic differentiation across large ranges. When range expansion occurs via serial founder effects, genetic drift will effectively be accelerated (Slatkin and Excoffier 2012), which can lead to the fixation and spread of rare alleles (Edmonds et al. 2004; Excoffier and Ray 2008; Excoffier et al. 2009). While the extent to which range expansion, in contrast to allopatric isolation, has shaped genetic diversity in wild populations is poorly known, several studies have identified signatures of population expansion related to Holocene range expansion. These include findings of increased spatial structure and shifts in allele frequency in the newly colonized range relative to the ancestral range (Graciá et al. 2013) and spatial sorting of mitochondrial lineages after expansion from a single refugium (Streicher et al. 2016).
Spatially-explicit coalescent simulation methods (Ray et al. 2010) have created unprecedented opportunities for addressing these issues by modeling complex historical phylogeographic scenarios. By coupling ecological niche models (ENMs) with these simulation approaches, biogeographic hypotheses can be generated and tested in a unified framework (Knowles et al. 2007; Richards et al. 2007). These coupled approaches can be used to test hypotheses regarding the relative effects of distance, habitat, and past climate (He et al. 2013; Massatti and Knowles 2016) in generating current genetic structure, thus refining understanding of nuanced historical patterns and processes.
Researchers employing spatially-explicit simulation procedures have in practice used a single climate reconstruction to hindcast species’ historical ranges (e.g., He et al. 2013; Massatti and Knowles 2016; Knowles and Massatti 2017). However there is considerable uncertainty regarding past climate, and modeling past distributions using alternate climate models or ENM thresholding rules can result in differing predictions regarding the location and extent of historic ranges and refugia (Waltari et al. 2007; Waltari and Guralnick 2009). Thus, different plausible climate reconstructions can indicate varying levels of allopatric isolation and range expansion. To address this, Alvarado-Serrano and Knowles (2014) recently reviewed use of ENMs in phylogeographic studies and stressed the importance of incorporating uncertainty regarding past distributions, but acknowledged the lack of an established method for doing so.
In this study, we used population-level and range-wide genetic data from the painted turtle (Chrysemys picta), as well as ecological niche modeling and spatially-explicit coalescent simulations, to elucidate the phylogeographic and demographic history of this taxon, and broader applications. Trans-continentally distributed species, such as the painted turtle, can be particularly informative for exploring the effects of broad-scale shifts in climate on biodiversity, as they often exhibit considerable genetic structure associated with historic shifts in range, as well as other ecological factors (Fontanella et al. 2008; Lougheed et al. 2013). Specifically, we compared current patterns of genetic diversity with data simulated under three refugial scenarios, based on alternate reconstruction of past climate, in a model-testing framework to examine the historical roles of glacial isolation (allopatry) and post-glacial range expansion. We used three different empirical datasets that differed in spatial sampling (population-level versus range-wide) and genetic sampling (mitochondrial DNA versus nuclear microsatellites) schemes to infer demographic parameters and the most likely refugial scenario. We also used cross-validation procedures to evaluate the accuracy of refugial hypothesis testing and parameter estimation for each sampling scheme. The approach taken here represents a promising framework for distinguishing the effects of isolation and range expansion and more accurately characterizing the historical forces structuring biodiversity.
Materials and methods
Study system
The genus Chrysemys is common and broadly distributed across North America, and exhibits both morphological and physiological trait variability across its range (Ernst and Lovich 2009). Traditional taxonomy has recognized one species (Chrysemys picta) subdivided into four subspecies (Ernst and Lovich 2009). More recent phylogeographic research (Starkey et al. 2003; Jensen et al. 2015) has identified the southern subspecies as a monophyletic group, resulting in the elevation of this taxon to species status as C. dorsalis (TTWG (Turtle Taxonomy Working Group) et al. 2017). We focus on the three northern subspecies of C. picta, and refer to these throughout as picta, marginata, and bellii (Fig. 1). These three subspecies display considerable genetic variability, including multiple mitochondrial clades exhibiting spatial structure across the range. However, past research using phylogenetic methods has been unable to come to a clear consensus regarding the historical processes that have generated this variability, as there is a lack of strict monophyly among morphologically defined subspecies (Starkey et al. 2003; Jensen et al. 2015; Figure S1).

Map showing current range of Chrysemys picta with sampling sites for this study. Blue lines indicate approximate boundaries for traditional subspecies, with variation in morphology among species shown for three representative individuals (C. picta picta from Orange County, NY; C. picta marginata from Walworth County, WI; C. picta bellii from Buffalo County, WI). Black dots indicate sampling sites for the range-wide mitochondrial DNA dataset, with regional groupings delineated by dashed lines and labeled with numbers. Red diamonds show locations for populations-level datasets. Four hypothesized refugial locations are shown: one on the East Coast (EC), one on the Gulf Coast (GC), one in the Southwestern US/Mexico (SW), and one broad southeastern refuge encompassing the East and Gulf Coasts (SE)
Field procedures and sampling
Using permitted procedures, we obtained genetic samples from ongoing studies covering the full longitudinal breadth of the C. picta range, including all three northern subspecies. We obtained blood or tissue samples from two C. p. picta populations on Staten Island, NY (FK and LP) as well as from C. p. bellii (NE) and C. p. marginata (IN). All blood samples were collected via the dorsal coccygeal vein or brachial artery. Tissue samples from an additional C. p. picta site in mainland NY (BR) were obtained from the Ambrose Monell Cryo Collection at the American Museum of Natural History (AMNH). DNA extractions from blood and tissue were performed using a DNEasy Blood and Tissue Kit (Qiagen, Inc., Valencia, CA, USA).
We obtained DNA extracts from additional populations in Wisconsin (WI) and British Columbia (BC). Field and lab methods are described in Reid and Peery (2014; for WI) and Jensen et al. (2014; for BC). These samples were grouped based on previous population clustering analyses conducted for these regions using a location prior (Jensen et al. 2014; Reid et al. 2016). Wisconsin samples were taken from an eastern population (WIe) located in a zone of morphological intergradation between C. p. bellii and C. p. marginata, and a western population (WIw) in the traditional range of C. p. bellii. BC individuals were also taken from two localities assigned to separate clusters: the Thompson-Okanagan cluster in south-central BC (BCs) and the Sunshine Coast/Gulf Island/Mid-Vancouver Island cluster in western BC (BCw).
Microsatellite genotyping
Twelve polymorphic microsatellite loci were genotyped at the AMNH using primers available in the published literature (Table S1). PCRs were carried out on an Eppendorf Master cycler following established protocols, and all reactions used an annealing temperature of 58 °C. Duplicate samples were included on plates and used as a quality check.
Microsatellite primers are known to cross-amplify well in turtle species, likely due to genome conservatism (Engstrom et al. 2007), and most of the primers used here were developed in other chelonian groups. We carried out multiple quality control steps to ensure that these markers performed reliably and according to the assumptions of downstream analyses (i.e., no null alleles, minimal linkage, and adherence to Hardy-Weinberg equilibrium). Microsatellite genotypes were manually called and scored using GENEIOUS version 8.1.2 (Kearse et al. 2012). For consistency and reliability, all genotypes were scored independently by two observers and then compared. Genotypes were coded as missing if the data were unclear. Samples with missing data were eliminated until there was no more than 5% missing data for each population. After this initial screening, MICROCHECKER 2.2.3 (Van Oosterhout et al. 2004) and GENALEX 6.5 (Peakall and Smouse 2012) were used to check for genotyping errors. We tested each locus for departures from Hardy-Weinberg and linkage equilibria at each sampling site using exact tests as implemented in GENEPOP 4.2 (Rousset 2008). In both cases, we corrected for Type I error rates using the sequential Bonferroni procedure (Rice 1989).
Mitochondrial DNA sequencing
We amplified a 662-basepair fragment of the mitochondrial control region using previously published primers and PCR conditions (Starkey et al. 2003). Amplification was confirmed using gel electrophoresis. PCR products were then either directly sent for sequencing to Genewiz (South Plainfield, NJ, USA) or purified using Exosap (Affymetrix, Santa Clara, CA, USA) and then sequenced at the AMNH. Each sequence was aligned to published haplotypes (Starkey et al. 2003; Jensen et al. 2015) using GENEIOUS version 8.1.2 (Kearse et al. 2012). Sequences that did not match any previously published sequence were first searched using NCBI BLAST to confirm that these sequences were novel. Individuals exhibiting novel haplotypes were then resequenced for confirmation.
Range-wide mitochondrial dataset
To broaden our analysis we assembled a range-wide dataset of published mitochondrial control region sequences (microsatellite or other data were not available range-wide). We refer to this dataset hereafter as “mtDNA-Range”. For the mtDNA-Range dataset, we added the sites sampled for mitochondrial control region in Jensen et al. (2015) and thinned any sites with more than ten samples to ten. This resulted in a dataset with a similar number of sequences (n = 290) distributed over a much larger number of sites (90) compared to the population-level dataset. We grouped samples in the mtDNA-Range dataset into nine spatially contiguous regional samples (Fig. 1), and calculated summary statistics based on these groupings in order to maintain a similar number of statistics relative to the population-level datasets.
Population genetic diversity
We calculated population-level diversity statistics for microsatellite and mtDNA datasets using ARLSUMSTAT (Excoffier and Lischer 2010). For both microsatellite and mtDNA datasets, we used the number of alleles (K) averaged over all loci as a basic metric of diversity. We also calculated average heterozygosity (H) for microsatellite data and average pairwise nucleotide diversity (π) for mtDNA.
Genetic structure, and isolation by distance
The model-based clustering method implemented in STRUCTURE 2.3.4 (Pritchard et al. 2000) was used to evaluate range-wide population structure using the microsatellite data. Run length was set to 1,000,000 MCMC (Markov chain Monte Carlo) iterations after a burn-in period of 500,000 using correlated allele frequencies under an admixture model with no prior location information. The most likely number of clusters was determined by varying the number of clusters (K) from 1 to 15 with 10 independent runs per value of K, and calculating ∆K (Evanno et al. 2005) as implemented in Structure Harvester (Earl and vonHoldt 2012). To test for substructure, we grouped populations according to their cluster membership and repeated the above analyses on the reduced datasets. Results for the identified optimal values of K were summarized using CLUMPP (Jakobsson and Rosenberg 2007) and plotted using DISTRUCT v1.1 (Rosenberg 2004). We also calculated codominant genotypic distances for all pairs of individuals as well as mean distances for all pairs of populations and conducted principal coordinates analyses (PCoA) on both distance matrices in GENALEX.
We calculated pairwise FST for microsatellites and mtDNA using ARLSUMSTAT, which employs the AMOVA method described in Excoffier et al. (1992). We calculated the Pearson correlation between genetic distance and geographic distance using the base statistics package in R (R Core Team 2015). To evaluate the statistical significance of the relationship between genetic and geographic distance, we conducted a distance-based redundancy analysis (dbRDA) with the R package vegan (Oksanen et al. 2013) using X and Y coordinates of each population as predictors (after Kierepka and Latch 2016).
Demographic history
We constructed Bayesian skyline plots (BSPs) using the mtDNA-Range dataset in BEAST v.1.8.0 (Drummond and Rambaut 2007). The purpose of BSP analyses was twofold: to (1) estimate the magnitude and timing of population size changes in C. picta; and (2) identify a plausible mitochondrial DNA mutation rate consistent with glacial isolation and post-glacial expansion (Hoareau 2016). As population structure and overrepresentation of individual populations can affect inferences made using BSPs (Heller et al. 2013), we also thinned the mtDNA-Range dataset to create an additional dataset (mtDNA-Thinned) with a maximum of 2 individuals per site. Since the BSP analysis assumes panmixia, changes in the structure of the study population may influence the inferred timing of population size changes (Mazet et al. 2016). To consider the possibility that secondary contact after refugial isolation could be influencing BSP analyses, we constructed three additional datasets corresponding to samples taken from the ranges of the three northern Chrysemys subspecies (as defined in Jensen et al. 2015). We compared the resulting skyline plots and assessed congruence between parameter estimates for all datasets.
All BSP analyses were conducted using the HKY + gamma + I mutation model (after Jensen et al. 2015). Recent estimates of mutation rates are often higher than ancient rates (Ho et al. 2005). As such, to determine the rate most consistent with expansion after the Last Glacial Maximum (LGM), we assessed two per-lineage mutation rates (either a slow mutation rate of 1.75 × 10−8 or a faster, doubled mutation rate of 3.5 × 10−8 mutations/lineage/year) based on the published control region substitution rate of 1.75 × 10−8 substitutions/site/year estimated from the uplift of the isthmus of Panama for green sea turtles (Chelonia mydas; Formia et al. 2006). This approach is in line with the method proposed by Hoareau (2016) for calibrating mutation rates using the inferred timing of assumed demographic changes.
We reviewed, tested, and used mostly default priors for skyline plot analyses, with the exception being for skyline.popSize, where we used a wide uniform prior to account for the potential range of possible ancestral and current population sizes for different geographic subsets (initial value = 1 × 105, prior distribution = uniform (0.5 × 107). Chain length for each run was 100 million iterations, with the first 10 million chains discarded as burn-in, and chains sampled every 4000 iterations. The effective sample size (ESS) estimator was used to diagnose convergence, with ESSs > 200 for all parameters used as indicators of convergence (Drummond et al. 2006). Effective population sizes (Ne) were calculated from tau values output by BEAST by dividing by generation time (11 years, based on mean generation time estimates of 10.7–12.35 years in Wilbur 1975). We note, however, that the accuracy of the values reported for Ne could be affected by uncertainty in generation time and potential variability in generation time across the range for Chrysemys. The medians of the population size distributions at either the median date of the most recent common ancestor or at year zero were used as estimators of ancestral (NeA) and recent (NeR) population sizes, respectively. The 97.5 and 2.5% quantiles for posterior distributions were used to delineate confidence intervals.
Ecological niche modeling
We downloaded C. picta occurrence records from the Global Biodiversity Information Facility (GBIF) and VertNet (downloaded June 2016) and concatenated the results into a single occurrence dataset after removing duplicate localities. To minimize the effect of records potentially resulting from incorrect identification or introductions, we restricted the dataset to only those occurrences that intersected with the International Union for Conservation of Nature (IUCN) range polygon for C. picta (van Dijk 2011). To reduce the effects of spatial autocorrelation, we then spatially thinned the occurrence data with the R package spThin (Aiello-Lammens et al. 2015), which resulted in a dataset of occurrences with no neighbors less than 10 km away. We defined the background extent as the area underlying a minimum convex hull of the occurrence localities. The convex hull was buffered by one degree to avoid including areas potentially suitable for C. picta, but outside of historically occupied regions due to barriers to dispersal (Anderson and Raza 2010).
Models for current potential distribution were built using Maxent, a presence-background ecological niche modeling (ENM) method (Phillips et al. 2006). We included the 19 bioclimatic variables from WorldClim at 10 arcmin resolution, representing monthly climatic averages for current conditions, as predictors in our models (Hijmans et al. 2005). We then built multiple models with two independent datasets ranging from simple to complex, and identified an optimal model based on test data performance on spatial cross validation using the R package ENMeval (Muscarella et al. 2014). Model selection for Maxent done in this way often results in less complex models that use a subset of the input predictor variables (Merow et al. 2013). To more accurately evaluate cross-validation error given spatial structure in the environmental data used in ENM (Roberts et al. 2017), we employed a spatial partitioning strategy using the “block” method in ENMeval. To determine whether any areas in the predicted study extent (southern Canada, the United States, and northern Mexico) have non-analog environmental conditions compared to the areas used to train the model, we ran a multivariate environmental similarity surface (MESS) analysis (Elith et al. 2010) using the R package dismo (Hijmans et al. 2016).
We hindcasted our model to both the Last Glacial Maximum and the Mid-Holocene (MID) using the WorldClim bioclimatic datasets for these time periods, masked to our study extent. As different general circulation models (GCMs) can have starkly contrasting predictions for certain areas, we hindcasted to both LGM and MID using three GCMs available on WorldClim for both time periods (CCSM4, MIROC-ESM, MPI-ESM-P) and took the standard deviation to explore the variability among predictions. To identify potential discrete refugia, we applied a threshold calculated from the present-day occurrence data to each hindcasted LGM model. The threshold value chosen maximizes the sum of sensitivity and specificity (max SSS; Liu et al. 2013). The max SSS value was selected as a conservative threshold that minimized overprediction outside the IUCN range polygon and avoided bias by balancing overprediction and underprediction of suitable habitat in the past. To ensure that climate conditions in the hindcasted refugia did not strongly differ from those in the present-day training extent, we also ran MESS analyses on each GCM hindcast per time period to assess the degree of non-analog conditions with the present-day model background training extent.
Refugial hypothesis testing
To determine the best-supported model of glacial isolation and post-glacial range expansion in C. picta, we used spatially-explicit coalescent simulations coupled with approximate Bayesian computation (ABC) methods. We defined three alternate historical scenarios based on each GCM hindcast. To generate habitat suitability raster maps for simulating range expansion, we first reduced the resolution of our hindcasted potential distribution prediction grids with an aggregation factor of 5 (resulting in 25 arcmin resolution) to achieve a manageable number of total cells. For each model we generated a “refugial” raster in which all cells with habitat suitability scores greater than or equal to the max SSS threshold at the LGM and with >1 neighboring cells above this threshold were considered potential refugial habitat. We also generated an “expansion” raster, representing potential habitat through which C. picta may have dispersed in order to reach its current distribution, by averaging the LGM and mid-Holocene maps for a given climate model and considering all cells with averaged suitability > 0.1 to be potential habitat. We lowered the habitat suitability threshold for the expansion period to allow for dispersal through some areas separating potential past refugia from the current range that had low suitability values at all three time points considered (LGM, MID, and present), but were likely suitable at some time during the Holocene. Finally, we created a “contemporary” raster in which we assigned all cells within the current IUCN-defined species boundaries for C. picta as potential habitat. As inferred habitat suitability was relatively stable between the contemporary and mid-Holocene time points compared to the LGM to mid-Holocene (see below), we used this contemporary raster to simulate habitat suitability from the mid-Holocene onward.
Simulations were executed using SPLATCHE v.2.1 (Ray et al. 2010), which first performs a forward-time demographic simulation followed by a reverse-time coalescent simulation. The forward simulation involved three phases. For the first phase (“isolation”), beginning at the last interglacial (130,000 years ago; Dahl-Jensen et al. 2013) and ending at the LGM, only cells considered potential habitat in the refugial raster were considered suitable. For the second phase (expansion), beginning at the LGM and lasting until the mid-Holocene (6000 years ago), all cells coded as potential habitat in either the “refugial” or the “expansion” rasters were considered suitable. For the final (contemporary) phase, lasting from the mid-Holocene to the present, only cells coded as potential habitat in the “contemporary” raster were considered suitable. At the beginning of simulations, a single deme within each refuge was seeded with 1000 individuals each, after which populations were allowed to expand freely according to the parameters of the simulation. The per-generation population growth rate was set to 0.25 (corresponding to approximately 2% growth per year), with this growth rate based on a life table constructed for C. picta (after Wilbur 1975) and consistent with population growth rates reported by Frazer et al. (1991) in an expanding population that grew approximately 50% in two decades. All suitable cells in each phase were assigned the same carrying capacity (K) and migration rate (M) for a given simulation. This is a simplification from the iDDC approach that scales carrying capacity to habitat suitability (He et al. 2013). While modeled habitat suitability varied widely over the range of C. picta based on our ENMs, habitat suitability explained little variability in population densities reported in the literature for C. picta (Figure S2). We used the stochastic migration model in SPLATCHE, in which the number of emigrants from a given deme in a generation is drawn from a Poisson distribution centered around the number of individuals in that deme multiplied by the migration rate, and emigrants migrate randomly into one of the four adjacent demes, with no friction. To confirm that population size trajectories roughly matched those inferred from mitochondrial DNA data, we summed the number of individuals in all demes at 500-year intervals and compared the change in population size over time to the confidence intervals from BSP analyses.
For the coalescent simulations, a per-generation mutation rate of 1.925 × 10-7 was used for mtDNA, corresponding to the slower mutation rate used in BEAST analyses multiplied by 11 years per generation. For the microsatellite data, a mutation rate of 8 × 10-4 per generation was used based on a C. picta pedigree study (Pearse et al. 2002). The proportion of multi-step mutations was set to 0.22, corresponding to the average proportion observed over multiple pedigree studies (Peery et al. 2012). As SPLATCHE simulates haploid genes on the landscape rather than diploid individuals, simulated microsatellite genotypes were generated by combining alleles under the assumptions of Hardy-Weinberg equilibrium (Ray et al. 2010). As such, estimates of Ne and K made using microsatellite data here indicate the number of gene copies, rather than the number of diploid individuals. Sampling locations and number of samples at each site corresponded to the locations and number of samples in our observed population genetic dataset.
We conducted simulations using combinations of ten different carrying capacities (K, in terms of haploid individuals per deme; from 1000 to 10,000) and ten different migration rates (M, from 0.001 to 0.01) for each of the three refugial scenarios. The range of values for demographic parameters used in simulations was determined after exploring a broader range of parameters and evaluating the overlap between the distribution of genetic summary statistics (see below) for simulated and observed data using the gfitpca function in the R package ABC (Csilléry et al. 2012). One thousand coalescent simulations were conducted for each combination of migration rate, carrying capacity, refugial scenario, and sampling distribution resulting in 300,000 total simulations each for population-level microsatellites, population-level mtDNA, and range-wide mtDNA. Given the difficulties associated with projecting changes in range beyond the last interglacial period, a final step in which all lineages were combined into a single small deme (n = 200) for 1000 generations was included in these simulations to ensure coalescence during the last interglacial. To determine whether the possibility of deep coalescence would affect inferences made based on these simulations, we also conducted an additional set of simulations (ten for each combination of refugial scenario and sampling scheme) with a much larger population size (n = 10,000) and a much longer final duration (12,000 generations) during this final step. Intermediate values were used for migration rate (M = 0.005) and carrying capacity (K = 5000) in these simulations.
We used ARLSUMSTAT to calculate summary statistics for all simulated datasets as well as the observed datasets. We calculated 125 summary statistics for mtDNA and 120 summary statistics for microsatellites (Table S2). We then used parameter estimation and posterior probability functions in ABC to quantify support for demographic models. All estimates of demographic parameters were conducted using data from all simulations (i.e., not assuming a given refugial model), and as such, we averaged across uncertainty in refugial model selection when estimating demographic parameters. We conducted cross-validation to evaluate accuracy of parameter estimation and model selection using the rejection method as well as the neural net method. The neural net method utilizes nonlinear regression and importance sampling to improve inference and avoid loss of precision resulting from use of large numbers of summary statistics (the “curse of dimensionality”) as well as problems associated with correlations among variables (Blum and François 2017; Csilléry et al. 2010). We used cross-validation to determine the combination of estimation method and tolerance level that minimized error (after Beaumont et al. 2002). We conducted cross-validation of each method using three different tolerance levels (0.001, 0.01, and 0.05), as simple rejection methods are usually most accurate with smaller tolerance values, while neural net methods are less sensitive to choice of tolerance and often improve in accuracy with higher tolerance (Blum and François 2017). One hundred pseudo-observed datasets (PODS) were randomly chosen from the simulated data and used to estimate carrying capacity and migration using the cv4abc function, and 100 PODS from each of the refugial scenarios were classified using the cv4postpr function in ABC. Parameter estimation accuracy was evaluated as mean squared prediction error for K and M, and refugial scenario assignment error was evaluated as the frequency at which the true model for a given simulated dataset was not selected as the most likely model. We also estimated posterior probabilities for refugial scenarios for all datasets simulated using deep coalescences. Finally, we determined support for demographic models using the observed summary statistics as a target. We used posterior model probabilities to identify the best-supported model for each dataset, and Bayes factors to determine the level of support for each model (Kass and Raftery 1995).
Results
Population-level sampling and genotyping
Sample sizes for each site ranged from 18 to 40 (Table 1). One microsatellite marker (GmuD70) was discarded after initial genotyping revealed a high frequency of null alleles. Three of the eleven remaining microsatellite markers displayed significant departures from Hardy-Weinberg equilibrium in at least one population, as well as in the global test. We performed population structure analyses both with and without these markers and obtained similar results (Figure S3); as such, all analyses presented here use the full 11-locus dataset (referred to hereafter at “MSAT-Pop”). We identified four novel mitochondrial DNA control region haplotypes that had not previously been described; the sequences for these haplotypes were deposited in Genbank (accession numbers MH665354–MH665357). The final population-level control region dataset is referred to hereafter as “mtDNA-Pop”.
Genetic diversity
For MSAT-Pop, genetic diversity was lower in the western groups BCw and BCs (K = 4.45 and 6.45; H = 0.43 and 0.62; Table 1) compared to all other sites (K = 7.45 to 9.45; H = 0.67–0.77). For mtDNA-Pop, WIe exhibited the highest haplotype diversity (K = 5, π = 2.21), with other areas lower in diversity or completely monomorphic (K = 1 to 3, π = 0 to 0.79; Table 1). Diversity in the mtDNA-Range dataset showed a longitudinal pattern similar to the MSAT-Pop dataset, with higher diversity in the three easternmost zones (K = 7–14; π = 2.41–3.73) and declining diversity to the west (K = 3–5; π = 0.20–1.88).
Genetic structure and isolation by distance
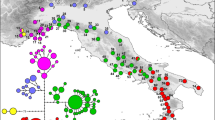
The Bayesian clustering analyses implemented in STRUCTURE for the MSAT-Pop dataset revealed strong evidence for K = 2 (∆K = 1780), with a secondary peak at K = 4 (∆K = 530). At K = 2, populations BR, FK and LP formed one cluster and all populations to the west were assigned strongly to a second cluster (Fig. 2). Re-analysis of this western cluster yielded a peak ∆K value at K = 3, which resulted in a clustering scheme almost identical to the scheme identified in the range-wide dataset for K = 4 (Fig. 2, Figure S3). At this value of K, IN clustered strongly with the two WI populations, NE formed a distinct cluster, and the two BC populations clustered together. Further analyses did not identify any substructure within the IN/WI group, but did identify the two BC populations as distinct clusters (Figure S3). PCoA showed similar results, and populations were arranged along the first principal component axis (explaining 63.6% of variability) from east to west (Figure S4).

Genetic clustering results based on microsatellite data for a two-population (K = 2) and four-population (K = 4) scenario for painted turtles. Barplots show individual ancestry coefficients for each individual, and pie charts on the map show approximate location of sampling sites and aggregated ancestry coefficients for each site based on K = 4. Distance between eastern sites is exaggerated to prevent overlap of pie charts (see Fig. 1)
Pairwise FST values for MSAT-Pop ranged from 0.02 to 0.34. FST exhibited a strong correlation with distance (r = 0.85; Fig. 3). Maximum FST values were higher for mtDNA-Pop (FST = 1 for several comparisons with no shared haplotypes), and reached an asymptote at much shorter distances (Fig. 3). dbRDA analyses indicated a significant association between location and genetic distance for both microsatellites (p = 0.002) and mtDNA (p = 0.002).

Pairwise genetic distance (FST) versus geographic distance for Chrysemys picta based on nuclear microsatellite (circles) and mitochondrial DNA data (triangles)
Demographic history
Effective sample size (ESS) values for nearly all parameters in BEAST analyses were >200, with the only exceptions being skyline group size parameters for the mtDNA-Range dataset. As skyline plots for the mtDNA-Range dataset were very similar to those obtained from the mtDNA-Thinned dataset (which did reach convergence), for simplicity we use the results for the mtDNA-Thinned dataset for range-wide demographic inferences.
Both the range-wide and subspecies datasets showed strong evidence for population expansion. The lower mutation rate (1.75 × 10−8/lineage/yr) gave results more consistent with demographic simulations (i.e., expansion shortly after the LGM and lower population sizes during the most recent glaciation; Fig. 4, Figure S5). The faster mutation rate resulted in a significant lag between glacial retreat and population expansion, as well as coalescence during the glacial period (data not shown). The magnitude of expansion ranged from 3.5-fold (for C. p. picta considered alone) to 18.5-fold (for the mtDNA-Thinned dataset). The estimated expansion period and most recent common ancestor (MRCA) were both more recent for the C. p. picta and C. p. bellii datasets than the range-wide dataset, while the estimated expansion and MRCA were earlier in C. p. marginata. However, confidence intervals for population size over time broadly overlapped for all three subspecies datasets and the range-wide datasets (Figure S5).

Bayesian skyline plots for Chrysemys picta showing population size on a log(10) scale through time, with the timing of the most recent glaciation shown above the plot. The 95% highest posterior density for population size from the range-wide analysis (all) is shown in grey
Ecological niche modeling
The concatenated occurrence dataset from GBIF and VertNet resulted in 3315 unique localities with coordinates, and removing those that did not intersect with the IUCN range polygon resulted in 3097 localities. Finally, spatial thinning by 10 km reduced this number to 1472 localities. The optimal ENM chosen used 13 of the 19 predictor variables and performed well on both the training dataset and an independent dataset of occurrence records (Figure S6). We projected the optimal model to the full study extent, and the highest suitability for C. picta ranged from the north-east to the central Atlantic coast of the U.S., westward to east of the Rocky Mountains, with pockets in New Brunswick and the Pacific coast of Mexico (which is likely outside the historical dispersal range; Fig. 5). The MESS results showed that only areas in western Mexico and Florida were considerably non-analog climatically with the model training area (Figure S7).

Present-day projected potential distribution for Chrysemys picta in North America based on ecological niche modeling of current occurrence data with bioclimatic predictors. Sampling sites and the range boundary for the species (white dotted line) are also shown
The highest agreement among GCMs for the LGM was for high suitability on the south-eastern Atlantic coast northeast of present-day Florida, and there was some agreement (CCSM4 and MPI-ESM-P) for high suitability on the Gulf coast between present-day Texas and Florida (Fig. 6; Figure S7). Across GCMs, the most variability was along the Gulf coast. The most non-analog areas across all GCMs were outside of potential refugia in the north-east U.S. and southern Canada (Figure S7). For the mid-Holocene, CCSM4 and MPI-ESM-P showed very similar patterns to the present, while MIROC-ESM showed possible range expansions to the south, west, and north (Figure S7).

Hindcasted potential distribution for the Last Glacial Maximum based on the current ecological niche model for Chrysemys picta and three different general circulation models. The dark lines indicate possible glacial refugia based on the max SSS threshold
Demographic parameter estimation and refugial hypothesis testing
The max SSS threshold value based on current occurrence data was 0.393. Applying this threshold to the three LGM hindcasts resulted in three distinct refugial scenarios: an East Coast-only scenario (EC) for the MPI-ESM-P model; a scenario with one large Southeastern refuge spanning the East Coast and Gulf Coast as well as a smaller refuge in the Southwest (SE + SW) for the MIROC-ESM model; and a scenario with distinct East Coast, Gulf Coast, and Southwest refugia (EC + GC + SW) for the CCSM4 model (Fig. 6). We refer hereafter to the simulations conducted based on climate hindcasts by these three different refugial configurations. These scenarios resulted in different numbers of demes occupied at the LGM during the demographic simulations, ranging from a low of 3 demes for the EC scenario to 42 demes and 108 demes for the SE + SW and EC + GC + SW scenarios, respectively. For all three scenarios, more demes were occupied in the warmer mid-Holocene (1027–1147 demes) than at present (828–833 demes).
For both microsatellites and mitochondrial DNA, cross-validation error associated with estimation of migration rate and carrying capacity, as well as misclassification of refugial models for PODS, was generally lowest for the neural net method and a tolerance level of 0.05 (Table S3a). Parameter estimates for migration and carrying capacity based on microsatellites were most accurate, and those based on the range-wide mitochondrial dataset were the least accurate. Refugial scenario assignment error using the neural net method and a tolerance of 0.05, on the other hand, was lowest for the range-wide mitochondrial dataset (2% of PODS incorrectly classified), although assignment error was also fairly low for microsatellites (7% of PODS incorrectly classified) and for the population-level mitochondrial dataset (12% of PODS incorrectly classified; Table S3a). Refugial scenario mis-assignments were mostly between two-and three-refugia scenarios; PODS generated using the EC model were almost never assigned to two-or three-refugia models, and vice versa (Table S3b). For the simulated datasets conducted using deep coalescences, assignment accuracy was still high for microsatellites (90% accurate), but lower for the population-level and range-wide mitochondrial sampling schemes (53.3% accurate and 67.7% accurate, respectively). This low accuracy was mainly due to mis-assignment of datasets simulated using the three-refuge-scenario (Table S3c).
ABC estimates for migration and deme carrying capacity based on the observed mtDNA-Pop dataset were both low (K = 1568, 95% CI 0 to 4896; M = 1.3 × 10−3, 95% CI 3 × 10−4 to 2.6 × 10-3; Table 2, Figure S8). The inferred carrying capacity based on the mtDNA-Range dataset was higher (K = 6430, 95% CI 4735 to 8322), and migration rates were somewhat higher as well (M = 3.8 × 10−3; 95% CI 1.5 × 10−3 to 6.8 × 10−3), although confidence intervals overlap with those from the mtDNA-Pop dataset. The estimate for carrying capacity based on the MSAT-Pop dataset was almost identical to the estimate based on the mtDNA-Pop (K = 1479; 95% CI 66–4280). However, the microsatellite-based estimate for migration was substantially higher than the mitochondrial estimate (M = 8.7 × 10−3; 95% CI = 6.46 × 10−3 to 1.144 × 10−2; Table 2, Figure S8). All three datasets identified the EC refugial model as having the highest posterior probability. For the MSAT-pop and mtDNA-Range datasets, posterior probability for this model was high (0.94), and Bayes factors identified this model as strongly supported (BF > 20 for all model comparisons). For the mtDNA-Pop dataset, the posterior probability was lower (0.58) and Bayes factors were <20 for model comparisons. There was also support for the EC + GC + SW model (posterior probability = 0.379), and support for this model relative to the EC model was equivocal according to Bayes factors (BF = 1.516).
Discussion
Testing refugial hypotheses with spatially-explicit population genetic models
Phylogeographic methods and ENMs provide complementary insights into changes in the distributions of species in response to past climatic events. Past work comparing ENM hindcasts of refugia to inferences from phylogeography has found broad agreement across many taxa (Waltari et al. 2007; Rödder et al. 2013). However, these comparisons are often qualitative in nature, using niche modeling as a secondary or parallel line of evidence (e.g., Buckley et al. 2010; Beatty and Provan 2010). The analytical framework described here follows the integrative distributional, demographic and coalescent (iDDC) approach described by He et al. (2013). This provides a means of explicitly linking alternative predictions from ENMs to genetic simulations, in order to test which potential refugial configurations and demographic parameters related to expansion best explain the current distribution of genetic variability.
Using the iDDC approach, Knowles and Massatti (2017) also identified range shifts (as opposed to isolation) as a key force in structuring genetic diversity in a grasshopper inhabiting a sky-island system. Our approach differs from the latter in several key respects. Most importantly, Knowles and Massatti’s isolation-only scenario used a static map of habitat suitability for the entire period being simulated, while all of our alternate refugial hypotheses incorporated both isolation and dynamic shifts in range via changes in habitat suitability over time. Our ENM results indicated large spatial shifts in habitat suitability over time for C. picta, such that there was almost no overlap between suitable habitat at the LGM and suitable habitat at the mid-Holocene and present time points. Consequently, incorporating range shifts in all historical models is justifiable and necessary, both in this situation and likely for similar analyses involving other widespread species inhabiting previously glaciated areas.
Another key difference in the analytical framework employed here is the use of alternate GCMs to parameterize potential refugial scenarios. Hindcasting ENMs to identify suitable habitat in the past includes many potential sources of error, including GCM choice. Using multiple models can help account for this source of error and identify plausible past distributions given model uncertainty. In light of our results, we believe that multiple GCMs should be used to guide the formulation of alternate hypotheses regarding past distributions. While other sources of error in hindcasting exist, we note that the three refugial scenarios used here capture in large part the variability among GCMs in hindcasted habitat suitability, including variability in the existence and extent of suitable habitat in the Gulf Coast and in the West. The best-supported historical scenario included a refuge in the area with higher overall stability across model predictions (the East Coast).
We evaluated three genetic and spatial sampling schemes for inferring past refugial scenarios, each of which potentially contained different kinds of information. Compared to mitochondrial DNA sequence data, microsatellites provide the advantage of multiple rapidly-evolving independent nuclear loci, but do not retain as much genealogical information. In our analyses, the mtDNA-Range dataset provided the greatest accuracy in terms of correctly assigning PODS to refugial scenarios, but accuracy was sensitive to the possibility of deep coalescences. The accuracy of inferences made using the MSAT-Pop dataset, on the other hand, was slightly lower, but less sensitive to coalescence time. As all three genetic datasets supported the EC scenario as the most likely of the three, expansion from a single refugium seems to be the most plausible scenario for the history of Chrysemys in the late Pleistocene. However, the uncertainty associated with inferences made using mitochondrial DNA datasets and the lack of deeper genealogical information inherent in microsatellite data leave open the possibility that genetic diversity in Chrysemys may also have been shaped by more ancient historical events, such as changes in range and population size occurring before the last interglacial period, that are not reflected by microsatellite data.
We did not include samples from the southern painted turtle (Chrysemys dorsalis) in this analysis for multiple reasons, the most practical being the lack of available population-level data for this species. Additionally, based on a previous analysis of hindcast range shifts in turtles over the Quaternary (Rödder et al. 2013), ENMs predict very little suitable habitat for this species during glacial episodes, with zero predicted suitable habitat for the LGM. This result may be influenced by this species’ relatively small current range and the artificial truncation of the climate envelope occupied by C. dorsalis at its southern range border at the Gulf of Mexico. All the same, the inability to accurately model a refugial range for this taxon at the LGM effectively prevents its inclusion in the framework used here. The C. picta and C. dorsalis mitochondrial clades are spatially distinct and likely split well before the LGM, and likely before the Quaternary itself (2.7–3.47 million years ago; Starkey et al. 2003). Although there may be some introgression of C. dorsalis nuclear genes into C. picta, or vice versa, where ranges of the two taxa overlap (Jensen et al. 2015), we believe that the exclusion of C. dorsalis did not significantly bias the results of this study, as populations used for the microsatellite datasets were far north of this overlap and all three datasets converged on the same refugial hypothesis. However, future studies including C. dorsalis could be useful in determining the extent of gene flow among these species after range expansion.
Isolation and expansion as drivers of variability in widely distributed taxa
Like many widespread taxa, Chrysemys picta exhibits genetic and morphological variability across its range. Allopatric isolation in glacial refugia has traditionally been invoked for generating the spatial segregation of this variability across painted turtles (Bleakney 1958) as well as numerous other species (Hewitt 2000). While C. picta morphotypes have traditionally been interpreted as resulting from three or more lineages that arose in allopatry (Bishop and Schmidt 1931; Bleakney 1958), our nuclear genetic data were more consistent with expansion from a single refugium. This result suggests that genetic differentiation during range expansion and isolation-by-distance are a more likely explanation for current genetic variability than historical isolation in multiple refugia in this species. This finding reinforces precautions against using morphological variability as a marker of allopatric isolation in widespread taxa (Byun et al. 1997). We do not reject subspecies groupings outright, as some traditional groupings may still delineate genetically distinct groups; however, our results suggest a stronger potential role for range expansion, rather than allopatry, in generating genetic structure for widely distributed taxa such as Chrysemys picta.
The alternative explanation of genetic and morphological differentiation via serial founder effects during spatial expansion of a diverse source population has received support in a variety of animal taxa, including sea snails (Acanthinucella spirata; Hellberg et al. 2001), black bears (Ursus americanus; Byun et al. 1997), and white-footed mice (Peromyscus leucopus; Ledevin and Millien 2013). These serial founder effects occur when the impacts of genetic drift outweigh the effects of gene flow during range expansion (Slatkin and Excoffier 2012). As such, species with low effective population sizes and low dispersal rates (such as turtles) may be particularly good candidates for experiencing serial founder effects. Indeed, founder effects have been identified as key to generating patterns of genetic diversity observed in spur-thighed tortoises (Testudo graeca) after colonization of the Iberian Peninsula (Graciá et al. 2013).
Our examination of phylogeography via spatially-explicit simulations identified a number of factors that likely structure current genetic diversity in taxa exhibiting recent range expansions. Declines in genetic diversity on the wavefront of expansion are a hallmark of range expansion (Edmonds et al. 2004). In all simulated scenarios, genetic diversity for both mitochondrial DNA and microsatellites was lower for populations at the western edge of the range (British Columbia), with the most extreme declines associated with scenarios incorporating lower carrying capacity and migration rates. Observed levels of genetic diversity and divergence in the western populations examined here were consistent with declines in diversity towards the edge of the range seen in other species, as predicted by the central-marginal hypothesis (Eckert et al. 2008).
Mitonuclear discord, in which patterns of mitochondrial genetic structure differ from patterns of nuclear genetic structure, has been observed in other expanding species and may be a result of stronger genetic drift and thus more severe serial founder effects in maternally-transmitted mitochondrial DNA (Streicher et al. 2016). Sex-biased dispersal patterns, which have been observed in many species (Greenwood 1980), could also contribute to mitonuclear discord. We did observe higher divergences and stronger isolation-by-distance in mitochondrial DNA compared to microsatellites in population-level analyses, though, the exact cause of this discord was unclear. Spatially explicit simulations supported somewhat higher dispersal rates for microsatellite data compared to mitochondrial data. Additionally, mitochondrial DNA did not show the expected pattern of lower effective population size compared to nuclear loci based on inferred carrying capacities. However, this theoretical expectation may be violated in natural populations due to greater skew in male reproductive success than female reproductive success or simply due to greater stochasticity inherent in mitochondrial DNA (Johnson et al. 2003; Ballard and Whitlock 2004). Taken together, these results provide some evidence for sex-biased dispersal rather than stronger founder effects as the cause of mitonuclear discord in this case. However, inferences of demographic parameters made using mitochondrial DNA data were generally less accurate than those made using microsatellite data, and as such the degree of sex-biased dispersal and the true causes of mitonuclear discord remain worthwhile avenues of investigation in this species.
We did not associate habitat suitability values with resistance to migration via the “Friction” parameter in SPLATCHE. Although landscape features, including the distribution of aquatic habitats, likely influence resistance to gene flow in this species (Reid et al. 2016), projecting these features into the deep past was beyond the scope of this analysis. Integrating the effects of changes in historical landscape as well as climate into simulations of range expansion would be useful for future phylogeographic investigations. Additionally, although we did not see a strong relationship between climate and population density, other landscape features (as well as biotic factors such as the presence of competitor species) could potentially have an effect on population size. Better characterizing the determinants of population density in Chrysemys and including these factors in simulations would also result in more realistic simulations of range expansion.
Finally, Chrysemys displays variability in many traits across its range. Intraspecific variability in body size and clutch size are independent of morphological subspecies definitions and are mostly related to latitude and elevation instead (Iverson and Smith 1993; Lindeman 1997). These patterns of variability also run counter to the longitudinal patterns of neutral genetic variability related to range expansion observed in this species. The latitudinal cline in body size in particular may reflect local adaptation to temperature (Lindeman 1997) and suggests a strong role for selection (in addition to the neutral processes of isolation and expansion examined here) in structuring geographic variability. Investigating patterns of adaptive genetic variability using a spatially-explicit framework similar to the one used here, would greatly improve our knowledge of how neutral and selective forces interact to produce the full range of variability observed in expanding populations. Genomic resources are now available for C. picta (Shaffer et al. 2013), making this species an ideal model for future investigations of the genetic basis of local adaptation in widespread taxa.
Data availability
Novel control region haplotypes have been uploaded to Genbank (accession numbers MH665354–MH665357). Microsatellite genetic data, animations showing range expansion dynamics for simulated scenarios, and R and Unix scripts used to perform ENM analyses and spatial simulations have been uploaded to the Dryad Digital Repository: https://doi.org/10.5061/dryad.8rb35rj.
References
Aiello-Lammens ME, Boria RA, Radosavljevic A, Vilela B, Anderson RP (2015) spThin: an R package for spatial thinning of species occurrence records for use in ecological niche models. Ecography 38:541–545
Anderson RP, Raza A (2010) The effect of the extent of the study region on GIS models of species distributions and estimates of niche evolution: preliminary tests with montane rodents (genus Nephelomys) in Venezuela. J Biogeogr 37:1378–1393
Alvarado-Serrano DF, Knowles LL (2014) Ecological niche models in phylogeographic studies: applications, advances and precautions. Mol Ecol Resour 14:233–248
Ballard JW, Whitlock MC (2004) The incomplete natural history of mitochondria. Mol Ecol 13:729–744
Beatty GE, Provan J (2010) Refugial persistence and postglacial recolonization of North America by the cold-tolerant herbaceous plant Orthilia secunda. Mol Ecol 19:5009–5021
Bishop SC, Schmidt FJW (1931) The painted turtles of the genus. Chrysemys Field Mus Nat Hist Publ Zool Ser 18:123–139
Bleakney S (1958) Postglacial dispersal of the turtle Chrysemys picta. Herpetologica 14:101–104
Blum MGB, François O (2017) Non-linear regression models for approximate Bayesian computation. Stat Comp 20:63–73
Buckley TR, Marske K, Attanayake D (2010) Phylogeography and ecological niche modelling of the New Zealand stick insect Clitarchus hookeri (White) support survival in multiple coastal refugia. J Biogeogr 37:682–695
Byun SA, Koop BF, Reimchen TE (1997) North American black bear mtDNA phylogeography: implications for morphology and the Haida Gwaii glacial refugium controversy. Evolution 51:1647–1653
Csilléry K, Francois O, Blum MGB (2012) ABC: an R package for approximate Bayesian computation (ABC). Methods Ecol Evol 3:475–479
Dahl-Jensen D, Albert MR, Aldahan A et al. (2013) Eemian interglacial reconstructed from a Greenland folded ice core. Nature 493:489–494
van Dijk PP (2011) Chrysemys picta. The IUCN Red List of Threatened Species. http://www.iucnredlist.org/details/163467/0. Accessed 13 February 2018.
Drummond AJ, Ho SY, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Earl DA, vonHoldt BM (2012) Structure Harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Edmonds CA, Lillie SA, Cavalli-Sforza LL (2004) Mutations arising in the wave front of an expanding population. PNAS 101:975–979
Elith J, Kearney M, Phillips S (2010) The art of modelling range-shifting species. Methods Ecol Evol 1:330–342
Ernst CH, Lovich JE (2009) Turtles of the United States and Canada, 2nd ed. Johns Hopkins University Press, Baltimore
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Ann Rev Ecol Evol S 40:481–501
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Excoffier L, Ray N (2008) Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol 23:347–351
Fontanella FM, Feldman CR, Siddall ME, Burbrink FT (2008) Phylogeography of Diadophis punctatus: extensive lineage diversity and repeated patterns of historical demography in a trans-continental snake. Mol Phylogenet Evol 3:1049–1070
Formia A, Godley BJ, Dontaine J-F, Bruford MW (2006) Mitochondrial DNA diversity and phylogeography of endangered green turtle (Chelonia mydas) populations in Africa. Conserv Genet 7:353–369
Frazer NB, Gibbons JW, Greene JL (1991) Growth, survivorship, and longevity of painted turtles Chrysemys picta in a southwestern Michigan marsh. Am Midl Nat 125:245–258
Graciá E, Botella F, Anadón JD, Edelaar P, Harris DJ, Giménez A (2013) Surfing in tortoises? Empirical signs of genetic structuring owing to range expansion. Biol Lett 9:20121091
Greenwood PJ (1980) Mating systems, philopatry and dispersal in birds and mammals. Anim Behav 28:1140–1162
He Q, Edwards DL, Knowles LL (2013) Integrative testing of how environments from the past to the present shape genetic structure across landscapes. Evolution 67:3386–3402
Hellberg ME, Balch DP, Roy K (2001) Climate-driven range expansion and morphological evolution in a marine gastropod. Science 292:1707–1710
Heller R, Chikhi L, Siegismund HR (2013) The confounding effect of population structure on Bayesian skyline plot inferences of demographic history. PLoS ONE 8:e62992
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978
Hijmans RJ, Phillips S, Leathwick J, Elith J (2016) Package “dismo”. R package.
Ho SYW, Phillips MJ, Cooper A, Drummond AJ (2005) Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol Biol Evol 22:1561–1568
Hoareau TB (2016) Late glacial demographic expansion motivates a clock overhaul for population genetics. Syst Biol 65:449–464
Iverson JB, Smith GR (1993) Reproductive ecology of the painted turtle (Chrysemys picta) in the Nebraska Sandhills. Copeia 1993:1–21
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jensen EL, Govindarajulu P, Russello MA (2014) When the shoe doesn’t fit: applying conservation unit concepts to western painted turtles at their northern periphery. Conserv Genet 15:261–274
Jensen EL, Govindarajulu P, Russello MA (2015) Genetic assessment of taxonomic uncertainty in painted turtles. J Herpetol 49:314–324
Johnson JA, Toepfer JE, Dunn PO (2003) Contrasting patterns of mitochondrial and microsatellite population structure in fragmented populations of greater prairie-chickens. Mol Ecol 12:3335–3347
Kass R, Raftery A (1995) Bayes factors. J Am Stat Assoc 90:773–795
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012) Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kierepka EM, Latch EK (2016) Fine-scale landscape genetics of the American badger (Taxidea taxus): disentangling landscape effects and sampling artifacts in a poorly understood species. Heredity 116:33–43
Knowles LL, Alvarado-Serrano DF (2010) Exploring the population genetic consequences of the colonization process with spatio-temporally explicit models: insights from coupled ecological, demographic and genetic models in montane grasshoppers. Mol Ecol 19:3727–3745
Knowles LL, Massatti R (2017) Distributional shifts-not geographic isolation-as a probable driver of montane species divergence. Ecography. https://doi.org/10.1111/ecog.02893
Lindeman PV (1997) Does life-history variation in the turtle Chrysemys picta have a subspecific component? J Herpetol 31:155–161
Liu C, White M, Newell G (2013) Selecting thresholds for the prediction of species occurrence with presence-only data. J Biogeogr 40:778–789
Lougheed SC, Campagna L, Dávila JA, Tubaro PL, Lijtmaer DA, Handford P (2013) Continental phylogeography of an ecologically and morphologically diverse Neotropical songbird, Zonotrichia capensis. BMC Evol Biol 13:58
Massatti R, Knowles LL (2016) Contrasting support for alternative models of genomic variation based on microhabitat preference: species-specific effects of climate change in alpine sedges. Mol Ecol 25:3974–3986
Mazet O, Rodriguez W, Grusea S, Boitard S, Chikhi L (2016) On the importance of being structured: instantaneous coalescence rates and human evolution—lessons for population size inference? Heredity 116:362–371
Merow C, Smith MJ, Silander JA (2013) A practical guide to MaxEnt for modeling species’ distributions: what it does, and why input settings matter. Ecography 36:1058–1069
Muscarella R, Galante PJ, Soley-Guardia M, Boria RA, Kass JM, Uriarté M, Anderson RP (2014) ENMEval: an R package for conducting spatially independent evaluations and estimating model complexity for MaxEnt ecological niche models. Methods Ecol Evol 5:1198–1205
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D et al. (2013) Package “vegan.” R package.
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research: an update. Bioinformatics 28:2537–2539
Pearse DE, Janzen FJ, Avise JC (2002) Multiple paternity, sperm storage, and reproductive success of female and male painted turtles (Chrysemys picta) in nature. Behav Ecol Sociobiol 51:164–171
Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Bëer E, Robinson S, Váaquez-Carrillo C, Pauli JN, Palsbøll PJ (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Model 190:231–259
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Provan J, Bennett KD (2008) Phylogeographic insights into cryptic glacial refugia. Trends Ecol Evol 23:564–571
R Core Team (2015). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
Ray N, Currat M, Foll M, Excoffier L (2010) SPLATCHE2: a spatially explicit simulation framework for complex demography, genetic admixture and recombination. Bioinformatics 26:2993–2994
Reid BN, Peery MZ (2014) Land use patterns skew sex ratios, decrease genetic diversity and trump the effects of recent climate change in an endangered turtle. Divers Distrib 20:1425–1437
Reid BN, Mladenoff DJ, Peery MZ (2016) Genetic effects of landscape, habitat preference and demography on three co-occurring turtle species. Mol Ecol 26:781–798
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Richards CL, Carstens BC, Knowles LL (2007) Distribution modelling and statistical phylogeography: an integrative framework for generating and testing alternative biogeographical hypotheses. J Biogeogr 34:1833–1845
Riddle BR (2016) Comparative phylogeography clarifies the complexity and problems of continental distribution that drove A.R. Wallace to study islands. P Natl Acad Sci USA 113:7970–7977
Rödder D, Lawing AM, Flecks M, Ahmadzadeh F, Dambach J, Engler JO et al. (2013) Evaluating the significance of paleophylogeographic species distribution models in reconstructing Quaternary range-shifts of Nearctic chelonians. PLoS ONE 8:e72855
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Sandel B, Arge L, Dalsgaard B, Davies RG, Gaston KJ, Sutherland WJ, Svenning J-C (2011) The influence of late Quaternary climate-change velocity on species endemism. Science 334:660–664
Shaffer HB, Minx P, Warren DE, Shedlock AM, Thompson RC, Valenzuela N et al. (2013) The Western Painted Turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol 14:R28
Slatkin M, Excoffier L (2012) Serial founder effects during range expansion: a spatial analog of genetic drift. Genetics 191:171–181
Starkey DE, Shaffer HB, Burke RL, Forstner MRJ, Iverson JB, Janzen FJ, Rhodin AGJ, Ultsch GR (2003) Molecular systematics, phylogeography, and the effects of Pleistocene glaciation in the painted turtle (Chrysemys picta) complex. Evolution 57:119–128
Streicher JW, McEntee JP, Drzich LC, Card DC, Schield DR, Smart U, Parkinson CL, Jezkova T, Smith EN, Castoe TA (2016) Genetic surfing, not allopatric divergence, explains spatial sorting of mitochondrial haplotypes in venomous coral snakes. Evolution 70:1435–1449
TTWG (Turtle Taxonomy Working Group), Rhodin AGJ, Iverson JB, Bour R, Fritz U, Georges A, Shaffer HB, van Dijk PP (2017) Turtles of the World. Annotated checklist and atlas of taxonomy, synonymy, distribution, and conservation status, 8th ed. Chelonian Research Foundation, Lunenburg, MA.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Waltari E, Guralnick RP (2009) Ecological niche modelling of montane mammals in the Great Basin, North America: examining past and present connectivity of species across basins and ranges. J Biogeogr 36:148–161
Waltari E, Hijmans RJ, Peterson AT, Nyári ÁS, Perkins SL, Guralnick RP (2007) Locating Pleistocene refugia: comparing phylogeographic and ecological niche model predictions PLoS ONE 2:e563
Wilbur HM (1975) The evolutionary and mathematical demography of the turtle Chrysemys picta. Ecology 56:64–77
Katalin Csilléry, Michael G.B. Blum, Oscar E. Gaggiotti, Olivier François (2010) Approximate Bayesian Computation (ABC) in practice. Trends in Ecology & Evolution 25:410–418
L. Lacey Knowles, Bryan C. Carstens, Marcia L. Keat (2007) Coupling Genetic and Ecological-Niche Models to Examine How Past Population Distributions Contribute to Divergence. Current Biology 17:940–946
David R. Roberts, Volker Bahn, Simone Ciuti, Mark S. Boyce, Jane Elith, Gurutzeta Guillera-Arroita, Severin Hauenstein, José J. Lahoz-Monfort, Boris Schröder, Wilfried Thuiller, David I. Warton, Brendan A. Wintle, Florian Hartig, Carsten F. Dormann (2017) Cross-validation strategies for data with temporal, spatial, hierarchical, or phylogenetic structure. Ecography 40:913–929
Ronan Ledevin, Virginie 2013 (2013) Congruent morphological and genetic differentiation as a signature of range expansion in a fragmented landscape. Ecology and Evolution 3:4172–4182
Beaumont MA, Zhang W, Balding DJ (2002) Approximate Bayesian Computation in population genetics. Genetics 162:2025–2035
Engstrom TN, Edwards T, Osentoski MF, Myers EM (2007) A compendium of PCR primers for mtDNA, microsatellites, and other nuclear loci for freshwater turtles and tortoises. Chel Res Mono 4:124–141
Acknowledgements
Blood and tissue samples were collected under the appropriate permits (IACUC permits from Earlham College and the University of Wisconsin-Madison; Indiana Scientific Purposes license #16-030; Scientific Collecting Permit #135 from the Wisconsin Department of Natural Resources; New York License to Collect and Possess #1757; UBC Animal Care Certificate # A11-0163 and BC Ministry of Environment sampling permit # VI11-71744). The Sackler Institute of Comparative Genomics and George Amato provided lab facilities and materials. Simulations and other computationally intensive analyses were carried out through the CUNY High Performance Computing Core, College of Staten Island, City University of New York. JBI was supported by the Earlham College Summer Research Fund. BNR was supported by a Gerstner Foundation postdoctoral fellowship and the Russ Gilder Graduate School at the American Museum of Natural History. Finally, comments provided by the editorial staff and three anonymous reviewers greatly improved the manuscript, and we thank them for their hard work and insight.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Reid, B.N., Kass, J.M., Wollney, S. et al. Disentangling the genetic effects of refugial isolation and range expansion in a trans-continentally distributed species. Heredity 122, 441–457 (2019). https://doi.org/10.1038/s41437-018-0135-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0135-5
This article is cited by
-
Meta-analysis reveals less sensitivity of non-native animals than natives to extreme weather worldwide
Nature Ecology & Evolution (2023)
-
Genome-wide genetic variation coupled with demographic and ecological niche modeling of the dusky-footed woodrat (Neotoma fuscipes) reveal patterns of deep divergence and widespread Holocene expansion across northern California
Heredity (2021)
-
Range-wide phylogeography of Blanding’s Turtle [Emys (= Emydoidea) blandingii]
Conservation Genetics (2019)
