
Abstract
Genomic data have the potential to inform high resolution landscape genetic and biological conservation studies that go far beyond recent mitochondrial and microsatellite analyses. We characterize the relationships of populations of the foothill yellow-legged frog, Rana boylii, a declining, “sentinel” species for stream ecosystems throughout its range in California and Oregon. We generated RADseq data and applied phylogenetic methods, hierarchical Bayesian clustering, PCA and population differentiation with admixture analyses to characterize spatial genetic structure across the species range. To facilitate direct comparison with previous analyses, we included many localities and individuals from our earlier work based on mitochondrial DNA. The results are striking, and emphasize the power of our landscape genomic approach. We recovered five extremely differentiated primary clades that indicate that R. boylii may be the most genetically differentiated anuran yet studied. Our results provide better resolution and more spatially consistent patterns than our earlier work, confirming the increased resolving power of genomic data compared to single-locus studies. Genomic structure is not equal across the species distribution. Approximately half the range of R. boylii consists of a single, relatively uniform population, while Sierra Nevada and coastal California clades are deeply, hierarchically substructured with biogeographic breaks observed in other codistributed taxa. Our results indicate that clades should serve as management units for R. boylii rather than previously suggested watershed boundaries, and that the near-extinct population from southwestern California is particularly diverged, exhibits the lowest genetic diversity, and is a critical conservation target for species recovery.
Similar content being viewed by others

Introduction
As we enter a new era of conservation genetics, it is increasingly clear that genomic-scale analyses of independent nuclear loci offer increased resolution for population genetic and phylogeographic studies by reducing statistical stochasticity associated with sampling a small number of coalescent gene genealogies (Rosenberg and Nordborg 2002). In some cases, applying a multilocus approach to the same taxa has led to major changes in our understanding of how lineages are structured in space and changed conservation management strategies compared to parallel single-locus analyses (Spinks et al. 2010; Myers et al. 2013). However, there are still relatively few examples of side-by-side comparisons of genomic and mitochondrial DNA (mtDNA) (with or without a small number of complementary nuclear loci), and a much larger sample of studies is needed to test specific hypotheses concerning how often large data sets lead to qualitatively different conclusions, rather than incremental increases in resolution. Both theory and limited data suggest that genomic approaches can lead to fundamentally different conclusions (Rosenberg and Nordborg 2002), but we need additional case studies to help establish guidelines for conservation managers to decide when a system should be revisited with genomic data. Here, we present a case study in a declining California amphibian.
Amphibian population declines are a global concern, driven by habitat degradation, climate change, emerging diseases, pesticides, introduced species, and pollution (Houlahan et al. 2000; Kiesecker et al. 2001; Stuart et al. 2004; Whiles et al. 2013). California has been a region of intense amphibian declines, with severe reductions across a wide range of anurans and urodeles (Hayes and Jennings 1986; Davidson 2004; Vredenburg et al. 2007; Kupferberg et al. 2012; Rogers and Peacock 2012; Ryan et al. 2014; Thomson et al. 2016).
One species that has received renewed attention from regulatory agencies is the foothill yellow-legged frog (Rana boylii), a species that historically was widely distributed from southwestern Oregon to southern California. This frog breeds exclusively in slow-flowing stream habitats and faces multiple threats, leading to its continued designation as a California Species of Special Concern (Thomson et al. 2016), its consideration for federal listing (US Fish and Wildlife Service 2015), and most recently its designation as a candidate for listing under the California Endangered Species Act in June 2017. The species is an important conservation target in its own right, and also serves as a sentinel species for stream ecosystems throughout its range. Several R. boylii studies have investigated the modification and loss of its stream breeding habitat through the construction of dams (Lind et al. 1996, 2016; Kupferberg et al. 2012), with one set of analyses showing that it has disappeared from at least 50% of historical localities (Davidson et al. 2002; Davidson 2004). Widely applied pesticides may be particularly problematic for R. boylii (Davidson et al. 2002; Davidson 2004; Bradford et al. 2011; Kerby and Sih 2015), and recent studies suggest that chytridiomycosis caused by the fungus Batrachochytrium dendrobatidis (Bd) constitutes a significant threat to the species in some areas (Ecoclub Amphibian Group et al. 2016; Adams et al. 2017). Experimental and field-based studies of R. boylii have also demonstrated (1) synergistic effects of Bd infection and pesticides on metamorphic growth (Davidson et al. 2007), and (2) that the combination of Bd infection, drought and invasive bullfrog (Rana catesbeiana) occurrence can affect Bd prevalence and load (Adams et al. 2017), suggesting that site and lineage-specific effects may be important drivers of decline, and equally important targets for recovery.
The combination of its status as a state-protected species, extreme habitat specialization, multiple, site-specific environmental challenges, and consideration for federal and state listing make information regarding the spatial genetic structure of R. boylii critical for biologically motivated recovery planning (McCartney-Melstad and Shaffer 2015). R. boylii also stands as a sentinel species for healthy stream ecosystems in California—stream flow manipulations have been identified as the primary cause of decline (Thomson et al. 2016), and a comparison of regulated and unregulated montane streams found that unregulated stream flows were associated with greater genetic diversity (Peek 2011).
A previous mtDNA study found evidence of spatial genetic structure across R. boylii, but had relatively low resolution and yielded sometimes puzzling results with respect to geography (Lind et al. 2011). Several results from that study were broadly concordant with those from other aquatic amphibians in California, including the discovery of unique, relatively differentiated populations in the extreme southeastern (Kern River drainage), southwestern (southern Monterey County), and the northern (Willamette River valley in central Oregon) localities and a very general concordance between drainage basins and genetic lineages, particularly with respect to the Sacramento-San Joaquin drainage system of central California. However, Lind et al. (2011) were potentially hampered by the low resolving power and idiosyncratic evolutionary history of a single mitochondrial marker, precluding definitive conclusions for molecular ecological and conservation decision making. We therefore applied a genomic restriction site-associated DNA sequencing (RADseq) approach to collect genetic variation for tens of thousands of nuclear loci for samples from across the range of the species, including many of the same samples and sites studied by Lind et al. (2011). Our goal was to provide a definitive analysis of genetic variation that would allow upcoming regulatory decision-makers to best evaluate R. boylii as a candidate species for listing across its range, and to directly compare the value of single-locus vs genomic inferences in the same study system.
Methods
Laboratory methods
Tissues from 93 R. boylii samples were obtained from 50 locations (between 1 and 9 samples per locality) in California and Oregon across the geographic range of the species (Santos-Barrera et al. 2004; IUCN et al. 2008) (Fig. 1, Table S1). Priority was given to samples representing each of the mtDNA clades recovered by Lind et al. (2011), with 47 of the samples in the present study drawn from 21 of the 34 localities used by Lind et al. (2011), and 30 biological samples overlapping between the two studies. DNA was extracted from tissues using a salt extraction protocol (Sambrook and Russell 2001). Dual-indexed sequencing libraries were prepared using the 3RAD protocol, a variant of the double-digest RADseq protocol (Peterson et al. 2012), with MspI as the common-cutting enzyme, SbfI as the rare-cutting enzyme, and ClaI as the dimer-cleaving enzyme (Hoffberg et al. 2016).

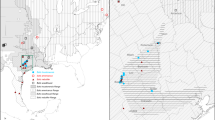
a Map of Rana boylii sample localities. County outlines are shown within California and Oregon. Light yellow indicates the IUCN range map of R. boylii, excluding a small disjunct population at Sierra San Pedro Martir, Baja California, Mexico. Dots are colored by major grouping as determined in the b RAxML phylogeny, with midpoint rooting. Nodes without dots received >95% bootstrap support, those with white dots received <95% but greater than or equal to 75% bootstrap support, while nodes with gray dots received <75% bootstrap support
Individual libraries were pooled at equimolar ratios and size selected in a single batch to between 500 and 700 bp using a Pippin Prep (Sage Science, Beverly, MA). Libraries were sequenced on two 100 bp paired-end HiSeq 2000 lanes. However, the second sequencing read for each fragment (R2) failed on one of these lanes, so we retained only the first reads (R1s) from both lanes to take advantage of the increased depth from two lanes of sequencing. For both lanes, custom sequencing primers were used that overlapped the restriction sites so that the first base of each read was the first base following the restriction site.
Quality control and clustering
To assemble the data set used for population genetic and phylogeographic analyses, pyRAD v3.0.66 (Eaton 2014) was run with the following parameters: #8/Mindepth = 10, #10/Wclust = 0.96, #11/Datatype = ddrad, #12/MinCov = 4, #13/MaxSH = 55, #24/MaxH = 2. These parameters correspond to a minimum depth of 10 to include a locus for a sample, a minimum clustering threshold of 96% similarity, a requirement that at least four samples have data to retain a locus, a maximum number of individuals being heterozygous for a particular base of 55, and a requirement that samples contain no more than two heterozygous sites at a locus to retain that locus. Mindepth and MinCov are filters for data quality and missingness, respectively, while MaxSH and MaxH serve as filters against potential paralogs being combined in a cluster. The clustering threshold value, a critical but often underappreciated parameter, was the subject of a separate set of analyses that will be presented elsewhere (McCartney-Melstad et al. 2017).
Data analysis
Phylogenetic analysis was conducted on a concatenated sequence alignment with a maximum of 50% missing data (i.e., retaining those loci achieving a minimum depth of 10 in at least 47 of 93 samples) using RAxML v8.2.8 (Stamatakis 2014). The best-scoring maximum likelihood (ML) tree was found using 20 ML searches with the GTRGAMMA model, and 100 rapid bootstrap searches were conducted to assess node confidence. We used midpoint rooting to visualize the resulting phylogeny.
Population structure was evaluated using fastStructure v1.0 (Raj et al. 2014). First, loci were retained that contained up to 80% missing data, since tests of population differentiation have been shown to perform relatively well at this level (Fu 2014, and see Figure S1). Then, to reduce the effects of physical linkage among variants, a single SNP was randomly chosen from each remaining RAD locus, leaving 38,520 biallelic SNPs for fastStructure analyses across all samples. Ten different random number seeds were used for K values between 1 and 12 to determine the most likely value for K to explain population structure among all 93 samples as determined by marginal likelihood. Population structure analyses often return the deepest hierarchical structure present in the data and ignore finer scale structure within the recovered clusters (Vähä et al. 2007; Janes et al. 2017). To ensure that these finer, more subtle levels of population structure were identified, samples were divided into groups based upon their initial fastStructure population assignment, fastStructure was rerun for each initial group for 10 seeds ranging from K = 1 to K = 12, and this process was repeated recursively for additionally discovered groups, stopping when any K value with the greatest marginal likelihood was K = 1 or equal to the number of sampling localities in the sample subset.
Principal components analysis (PCA) was performed using SNPRelate v1.6.4 (Zheng et al. 2012) on the same set of 38,520 biallelic SNPs with a single SNP per RAD locus and no more than 80% missing data. Principal components were plotted against one another to visualize patterns of genetic variation using R 3.4 (R Core Team 2017).
We used TreeMix v1.13 (Pickrell and Pritchard 2012) to model genetic drift among sample localities while explicitly accounting for admixture. TreeMix was run with 10 different random number seeds for between 0 and 8 added migration edges. Individuals from the same locality were pooled together into 50 “population” samples, and sample size correction was disabled, as 28 of the localities consisted of a single individual. SNPs were included in the TreeMix analysis if at least one sample from each locality had data (6004 SNPs total). Locality topologies and admixture arrows were compared between random number seeds, and we present random seeds with the highest likelihood for each number of migration edges. The three-population test (Reich et al. 2009) as implemented in TreeMix v1.13 was also used to test for gene flow among the five major groups identified by RAxML and PCA using the 26,240 SNPs that contained data for at least one individual per group.
Following Lind et al. (2011), we used AMOVA to assess the influence of hydrological boundaries in structuring R. boylii populations (Excoffier et al. 1992). Samples were grouped according to the Watershed Boundary Database (USDA-NRCS et al. 2016) into drainage basins (6-digit hydrological unit codes, Table S1). AMOVA was conducted across all 93 samples in Arlequin 3.5.2.2 (Excoffier and Lischer 2010) with the following hierarchical levels: (1) basin, locality, individual, and (2) five major phylogenetic clade membership, locality, individual. AMOVA was also conducted separately within each of the five major phylogenetic clades to discern how much variation within each group is a result of differences among localities vs within individuals. Individual pairwise sequence divergences were calculated using dnadist from Phylip v3.696 with JC69 distances (Jukes and Cantor 1969; Felsenstein 1989), and among-group divergences were calculated by averaging all of the individual-individual pairwise distances between group members. We also estimated Fst between the five major clades recovered by RAxML and between all sampling localities using the Weir and Cockerham (1984) weighted Fst measurement as implemented in SNPRelate 1.6.4 (Zheng et al. 2012), considering all polymorphic SNPs that contained data for samples in both populations in a comparison. Tajima’s D and nucleotide diversity (π) were calculated as averages across the set of RAD loci not missing any data respectively within each of the five major RAxML clades using VCFtools v0.1.15 (Danecek et al. 2011).
Results
A total of 394,983,404 single-end 100 bp sequence reads derived from 93 samples across two HiSeq 2000 lanes were generated for this experiment. Individual frogs received between 2,353,591 and 6,263,131 reads (mean = 4,247,133, stdev = 840,333). Reads with more than four low-quality bases (phred scores below 20) were discarded, reducing our data set to an average of 3,056,113 single-end reads per frog for analysis (min = 1,698,025, max = 4,560,049, SD = 606,497).
After processing in pyRAD, 106,000 loci were recovered that contained data for at least four samples (Table S2). Because missing data among individuals is often high in RADseq studies (Eaton 2014) including ours, we generated two subsets of our data. The first, for phylogenetic (RAxML) analysis, contained no more than 50% missing data for any locus, resulting in 25,569 loci (2,435,662 bp per individual, Table S2). The second, for the fastStructure and PCA analyses, had no more than 80% missing data across all samples, and consisted of 38,575 loci with at least one SNP.
RAxML
We highlight five main monophyletic groups identified in the RAxML tree that each have 100% bootstrap support (Fig. 1b) and are geographically cohesive (Fig. 1a). The deepest split in this tree is between coastal populations south of San Francisco Bay (the blue + purple clade in Fig. 1b) and the rest of the species’ range. Within the first of these clades, two reciprocally monophyletic groups stand out. The first (purple in Fig. 1; the “southwestern California” clade) is from two nearby localities in the Central Coast Range of California in southernmost Monterey County. The second group (blue in Fig. 1; the “western California” clade) consists of seven localities from Alameda, Santa Clara, Santa Cruz, San Benito, and western Fresno Counties in central California. It is bounded by the Salinas River Valley to the south and west and San Francisco Bay to the north, and occurs in both coastal and interior flowing drainages. For both lineages, the widespread extirpation of R. boylii from the central coast and southern California precluded testing the historical geographic range boundaries of these groups. The southwestern California clade, which as far as we know now only occurs at our sampling localities, is particularly problematic.
This inclusive group is sister to a clade consisting of all populations north of San Francisco Bay plus all Sierran R. boylii populations, and consists of three primary subgroups. The first (green in Fig. 1; the “eastern California” clade) consists of three widely spaced localities in west-flowing drainages on the east side of California’s Central Valley in Tulare, Calaveras, and eastern Fresno Counties. The second group (red in Fig. 1; the “northwestern California/Oregon” clade) is widespread, and our sampling consists of 33 localities extending from north of San Francisco Bay through western and central California into Oregon (including Marin, Sonoma, Solano, Lake, Colusa, Glenn, Mendocino, Tehama, Humboldt, Trinity, Shasta, and Del Norte Counties in California, and Curry, Douglas, and Linn Counties in Oregon). The final group (orange in Fig. 1; the “northeastern California” clade) consists of five localities between the eastern margins of the northwestern California/Oregon clade and the northern margin of the eastern California clade. In our sampling it is restricted to Nevada, Placer, Yuba, and Plumas Counties in California’s Sierra Nevada.
Based on midpoint rooting, RAxML recovered the southwestern California clade as sister to the western California clade. The eastern California clade was sister to a clade consisting of the reciprocally monophyletic northwestern California/Oregon and northeastern California clades (Fig. 1b). The relationships among these groups were fully supported (bootstrap values of 100), but many of the shallower nodes within the five main clades received lower support. Bootstrap support values within the different groups differed: 0 of 3, 1 of 9, 4 of 7, 7 of 16, and 29 of 48 nodes received less than 95% bootstrap support in the eastern California, northeastern California, southwestern California, western California, and northwestern California/Oregon clades, respectively (gray and white nodes in Fig. 1b). These relationships were robust to the uneven sampling present in our data set, as a similar analysis of a data set downsampled to be more even recovered the same results (Figure S2).
PCA and fastStructure
PCA broadly supported the phylogenetic results from RAxML. PC1 and PC2 (explaining 11.3% and 8.7% of the genomic variance, respectively) separated the southwestern California and western California clades from each other and all other samples, with the southwestern California cluster most isolated from all others (Fig. 2a). PC3 (7.1% of the variance) separated the eastern California samples from all others, while PC4 (3.9% of the variance) distinguished the northeastern California samples from the northwestern California/Oregon and eastern California samples (Fig. 2b). PCs 5 through 8 (3.2–2.4% of the variance) suggest further substructure within the five main clades. More specifically, PC5 and PC8 are axes of genetic variation within the northwest California samples, separating the localities in Oregon from those further south (Fig. 2c, d). Similarly, PCs 6 and 7 break the western California and eastern California samples into subgroups, respectively.

Principal components analysis. SW southwestern California clade, W western California clade, E eastern California clade, NE northeastern California clade, NW northwestern California/Oregon clade
Within-clade substructure was most finely characterized by hierarchical fastStructure analysis and was also broadly concordant with the RAxML tree. For each of the ten random seeds tested, K = 4 was the configuration that maximized the marginal likelihood and was also the number of model components used to explain structure in the data (Fig. 3). The western California and southwestern California clades formed their own respective fastStructure populations and showed no evidence of admixed individuals. The remaining 66 individuals clustered into two populations, with 50 individuals typically clustering as purely northwest California/Oregon-clade animals and 16 individuals forming the fourth cluster, consisting of all individuals derived from the eastern + northeastern RAxMl clades. This latter group sometimes showed a slight degree of northwest California/Oregon clade membership (Figure 3), although these samples were fully resolved into distinct groups in the second round of clustering (labeled 2A in Fig. 3). The individuals that exhibited detectable admixture among the different random number seeds belong to the northeastern California clade, occurring in Yuba, Placer, Nevada, and Plumas Counties in California. They also all belong to a single clade representing the sister group to the northwestern California/Oregon samples in the RAxML tree (Fig. 1b).

Hierarchical fastStructure results. Each number/letter combination represents a single fastStructure run and has a corresponding map with matching colors representing the samples in the fastStructure plot
Following this initial fastStructure run, major groups were then isolated and recursively subjected to fastStructure analyses until the highest marginal likelihoods were observed for K values equal to one or the number of sampling localities. The four groups identified by the first, global fastStructure analysis differed greatly in their individual substructure (Fig. 3, Table S3). For instance, the global fastStructure population consisting of the eastern California and northeastern California clades required four additional rounds of fastStructure to reach the stopping condition, yielding eight distinguishable groups (Table S4). The population corresponding to the northwestern California/Oregon samples, however, required only two iterations to reach the stopping conditions and yielded just three distinguishable groups despite consisting of more than three times as many samples (50 vs 16) and covering a much larger geographical area. Similarly, the western California clade yielded five distinguishable groups after three rounds of fastStructure, while the southwestern California clade yielded two clusters corresponding to the two sampling localities.
TreeMix
TreeMix recovered topologies similar to RAxML, with some interesting differences. Model likelihoods improved dramatically until five migration edges were added, and increased modestly thereafter (Figure S3). The best-scoring TreeMix tree with five migration edges is shown in Fig. 4, its residuals are shown in Figure S4, and all other runs are shown in Figures S5-S12 (for locality information, see Fig. 4b and Table S1). The western California and southwestern California clades always formed reciprocally monophyletic groups, and the long branch between these clades and the remaining samples was used to root all TreeMix topologies. Similarly, the eastern California localities always formed a monophyletic group and were always sister to the northwestern California/Oregon and northeastern California samples.

The five localities that make up the northeastern California clade always formed a monophyletic group. When no migration was modeled, this group was sister to all northwestern California/Oregon localities (Figure S5), but when at least one migration edge was added the northeastern California localities were nested within the northwestern California/Oregon localities (Fig. 4, Figures S6-S12).
The first five added migration edges occurred: (1) from the ancestor of the eastern California samples to an early branch in the northeastern California samples and (2) to the adjacent locality 32 (northwestern California/Oregon), (3) from the ancestor of the central Oregon localities to the southwestern Oregon/far northwestern California localities, (4) from the ancestor of eastern California locality 14 to northeastern California locality 29, and (5) between localities within the northwestern California/Oregon clade (Fig. 4). Three-population tests did not indicate that any one of the five major RAXmL clades consist of a mixture of any two of the other four groups (Table S5).
AMOVA, Fst, and Tajima’s D
Results from AMOVA show that 29.6% of the total genomic variation is partitioned among drainage basins compared to nearly twice as much (53.9%) among the five major phylogenetic clades (Table 1). Most of the residual variation in the drainage basin AMOVA is attributed to variation among localities within water basins (38.0% as opposed to 18.0% in the phylogenetic clade analysis). This suggests that the deep population structure recovered by RAxML, PCA, and fastStructure is not perfectly captured by membership among the different HUC6-level water basins in California and Oregon, as might have been expected for this stream-restricted anuran. When AMOVAs were run separately for the five major clades, the variation accounted for by differences among sampling localities varied widely (Table 2). Variation among localities was lowest for the southwestern California (22.97%) and northwestern California (28.87%) groups, and was highest for the eastern California group (64.65%).
Fst values were extremely high among the five major clades (Table 3). The lowest Fst was 0.312 for the comparison between northwest California/Oregon and northeast California, and the highest value was 0.794 between the southwestern California samples and the eastern California samples. The southwestern California samples were the most differentiated group from each of the other four groups (average Fst = 0.711). Plotting the Fst values of comparisons between localities in the same major clade against geographic distance revealed a strong pattern of genetic isolation by geographic distance within clades (black in Fig. 5). Fst values were generally at least twice as high for comparisons between localities from different major clades compared to those in the same major clade across the same geographic distance (Fig. 5), and these comparisons tended to be less affected by geographic distance than within-clade comparisons, as is expected among highly divergent evolutionary lineages.

Fst values calculated between sampling localities vs the geographic distances separating localities, with red dots for comparisons between major clades
Tajima’s D values were positive for all clades except for the large, relatively unstructured northwestern California/Oregon, which was −0.98 (Table S4), suggesting that this group may have uniquely undergone a recent demographic bottleneck/expansion (Tajima 1989). Nucleotide diversity values ranged from 0.0010 for the southwestern California samples to 0.0034 for northwestern California/Oregon (Table S4). Average sequence distances tended to be greatest when comparing groups to the southwestern California clade, although the western and eastern California clades were also very differentiated (Table 3).
Discussion
Our case study of R. boylii population genomics emphasizes three key findings. First, our RADseq data provides a much clearer, geographically structured picture of landscape differentiation than was previously possible with mtDNA. Second, the depth of population structure is often extraordinarily deep, surpassing that found in any anuran for which similar data are available. And third, the critically imperiled southern populations harbor the greatest genetic variation, and should be the target of particularly intense conservation efforts.
Our work joins a series of range-wide analyses across California that highlight some emerging phylogeographic patterns and trends, although few employ genomic-level data and range-wide sampling. Both at continental (O’Connell et al. 2017, using RAD data) and California (Rissler et al. 2006, with more limited data sets) scales, deep within-lineage splits consistently reflect physical barriers to gene flow (O’Connell et al. 2017), sometimes overlain with recent range expansions and contractions that differentially affect mitochondrial and nuclear gene tree distributions (Spinks et al. 2010, 2014; Bryson et al. 2016; Phuong et al. 2017). The deepest genetic split in our RADseq analysis of R. boylii is between coast range populations south of the Sacramento-San Joaquin River Delta (or equivalently, San Francisco Bay) and those north of the Delta including the Sierra Nevada (Fig. 1), and the Delta has been previously identified as a consistent phylogeographic barrier across a range of taxa and data sets (Calsbeek et al. 2003; Lapointe and Rissler 2005; Rissler et al. 2006). A virtually identical pattern of primary differentiation across the Delta to that in R. boylii was recovered for the riverine endemic western pond turtle Emys (Actinemys) marmorata/pallida complex based on 89 nuclear SNPs (Spinks et al. 2014) and a similar, but slightly southward-shifted break was identified in the mountain kingsnake Lampropeltis zonata (Myers et al. 2013) but not in the California newt Taricha torosa (Tan and Wake 1995; Kuchta and Tan 2006). Other studies have regularly identified the Sacramento-San Joaquin Delta as an important barrier to gene flow (for example, the California red-legged frog, Richmond et al. 2014), suggesting that the Delta constitutes a consistent phylogeographic barrier across a diverse range of species and life histories. Although there are certainly exceptions to this pattern, many are based on modest, single-gene data sets, and we look forward to future genomic-level studies that revisit the factors shaping California phylogeography and the conservation recommendations derived from those analyses.
Phylogeography and population genetics of a deeply structured riverine species
The central finding of this study was the presence of deep, geographically structured genetic subdivision in R. boylii across its range in California and Oregon. Although clear patterns of genetic differentiation were evident across many different spatial scales, at the coarsest, range-wide level, five deeply divergent genetic groups were present across phylogenetic (Fig. 1), ordination (Fig. 2), and Bayesian clustering (Fig. 3) approaches, even when explicitly modeling admixture (Fig. 4). Fst among these groups was extraordinarily high (Table 3), and currently stands as the highest that we are aware of for any anuran (Monsen and Blouin 2004). That such variation exists and was not apparent in an earlier mtDNA analysis emphasizes the importance of resampling with genomic tools, especially for declining species that may lose important genetic variation and local adaptation without immediate conservation actions.
The relationships among the five main groups were well resolved with one interesting exception. The northeastern California clade appears most closely related (RAxML with 100% bootstrap support, Fig. 1, and TreeMix, Fig. 4 and Figures S5-S12) and most similar (using PCA, Fig. 2) to the northwestern California/Oregon clade, but is more closely allied with the eastern California clade in fastStructure analyses. This may hint at a complex relationship of the northeastern California population with its neighbors to the south and west. Although the three-population test did not indicate that the northeastern California samples are the result of an admixed group between the eastern California samples and those from the northwestern California/Oregon clade (Table S5), TreeMix analyses did recover a signal of migration between ancestors of the eastern California and northeastern California localities (or their ancestors) for all analyses with added migration edges (Fig. 4, Figures S6-S12). These results suggest multiple possible conservation solutions for the northeastern California group: they are both less evolutionarily unique than the other four clades (and thus of lower priority) and they also represent a group with a history of admixture and thus may be a rich source of potential adaptive variation (and are therefore of higher priority). Additionally, if the northeastern California group has indeed received recent migrants from both the eastern California and northwestern California/Oregon groups, then these geographically intermediate localities could be important for maintaining long-range metapopulation dynamics between a large population (northwestern California/Oregon) and a smaller, more imperiled population in eastern California. These disparate interpretations invite more comprehensive whole-genome analyses to quantify the intensity of selection and variation at adaptively important loci as additional genomic resources become available for these large-genome animals.
At a finer resolution, there are indications of additional deep hierarchical population structure within these five groups. While fastStructure recovered distinct population clusters for each sampling locality in the eastern California and southwestern California clades, the 33 northwestern California/Oregon localities supported just three subunits, one of which spanned more than 50,000 km2. The western California localities were intermediate in this regard, with fastStructure recovering five populations across seven localities. Similarly, nucleotide diversity varied by more than a factor of 2 among the major phylogenetic clades, with the western California clade exhibiting a value half that of northwestern California/Oregon. Given the lack of phylogenetic resolution within the northwestern California/Oregon clade and its negative Tajima’s D, both of which suggest a recent population expansion, this high level of nucleotide diversity is consistent with either a range expansion from multiple source populations or some limited admixture from other areas. One concern is that these estimates of genetic diversity may be influenced by sample size (Subramanian 2016) or relative area sampled, as the two southwestern California localities are extremely close together. However, this does not fully explain the depressed π values in southwestern California; our nine samples from locality 2 (western California clade) and six samples from locality 50 (northwestern California clade) had π values of 0.0012 and 0.0022, respectively, which are both higher than the 0.0010 registered for the nine samples from two southwestern California localities. The southwestern California samples appear to be undergoing marked reductions in nucleotide diversity compared to other parts of the range (Table S4), presumably reflecting the extreme population and range reductions in the region. As has been repeatedly demonstrated in a variety of taxa (Frankham et al. 2017), population reductions and inbreeding depression are important concerns, and may well call for assisted migration among the few remaining populations of the southwestern California clade.
Comparison to Lind et al., 2011
Comparison of our results with a previous, primarily mtDNA study of many of the same localities and individuals provides important insights into the increased utility of genome-level SNP data compared to the single-locus analyses that characterized earlier phylogeographic and conservation genetic analyses. Both Lind et al. (2011) and the current study recovered the genetic distinctiveness of populations at the periphery of the species range in southern Monterey (our southwestern CA) and Kern Counties (part of our eastern California) and in the central Sierra of California (clade C in Lind et al. 2011). However, the earlier analyses based on 1525 bp of mtDNA and 517 base pairs of a single nuclear intron indicated that range-wide variation in R. boylii was modest and those data had virtually no statistical power to resolve groupings of populations into more inclusive lineages.
In the current study, however, most of the ambiguities in both group membership and their relationships are resolved and clarified: major clades are well-supported, and the substructure that so clearly dominates Sierran and coastal populations south of San Francisco Bay are equally strongly supported. Other biogeographic details critical to both evolutionary understanding and effective management also emerged: our new data demonstrate that the weak evidence for a recent “trans-Central Valley leak”, as was recently (and reasonably) suggested by Richmond et al. (2014) based a single sample (HBS 37260), can be conclusively interpreted as a case of incomplete lineage sorting of mtDNA, and the confusing placement of samples from northwestern California (del Norte, Humboldt, and Lake Counties) or southwestern Oregon (Curry County) in Lind et al. (2011) resolves unambiguously in our study. Finally, one of the major results from Lind et al. (2011) was that genetic diversity in R. boylii was largely partitioned by hydrological boundaries, accounting for roughly 40% of the observed genetic variation. While this signal was also present in the current analysis, we recovered a lower proportion of variance described by water basins (30%), while a more informative partitioning by phylogenetic clade explained 54% of the total genetic variation. This may be partially be due to our use of USGS hydrological units as opposed to the hydrological regions employed by Lind et al. (2011), but the deep divisions and extraordinary Fst values for our genomic-level data indicate that clade-level historical divergence is the most important component of intraspecific differentiation in R. boylii.
Conservation of an ecological specialist
Field ecology and natural history studies have provided an essential foundation for conserving remaining populations of R. boylii and aiding in its recovery, documenting reasons for local declines (Lind et al. 1996, 2016; Davidson et al. 2002; Davidson 2004; Bradford et al. 2011; Kerby and Sih 2015) and susceptibility to pathogens and invasive species (Davidson et al. 2007; Ecoclub Amphibian Group et al. 2016; Adams et al. 2017). Landscape genomics, when conducted in the context of complete sampling and careful analyses, can add important historical and landscape-level insights that are absent from these studies, and help guide management and listing decisions for declining species. Our single strongest recommendation is to manage each of the five major phylogenetic clades (Fig. 1) identified herein as independent recovery units of R. boylii. The extraordinarily high Fst values among major clades are consistent with the interpretation that R. boylii is deeply divided taxon that demands conservation actions following a clade-level, in addition to a watershed-level, approach. The among-clade and among-locality Fst values observed here were considerably higher than those found over similar distances in the related Rana cascadae by Monsen and Blouin (2004), which has previously served as an exemplar of extremely strong genetic differentiation in an amphibian species. While major hydrological boundaries explain a reasonable portion of the broad-scale genetic diversity of R. boylii, there are also regions where distantly related samples occur in the same HUC6-level hydrological unit. This is particularly true in north-central California, where the northeastern California clade and some northwestern California/Oregon clade localities co-occur. Within these five units, our hierarchical analyses of population structure, visualized in Fig. 3, indicate that additional identifiable structure exists, sometimes down to the level of individual sites. Although our sampling is geographically quite complete, additional analyses that fill in gaps within and between drainages is necessary to fully identify management subunit boundaries that should help guide management decisions including potential dam removals (Lind et al. 1996, 2016).
Focus protection and recovery on the southwestern California unit
The southwestern California samples were the most genetically distinct of all the samples analyzed here, driving PC1 and PC2 in Fig. 2 despite consisting of just nine samples from two nearby localities. We recovered the lowest nucleotide diversity (π) in the southwestern California samples among the five major clades, and the two sampling localities in this clade separated into two distinct populations in fastStructure analyses despite a distance between sites of only 7 km. This contrasts sharply with the bulk of the northwestern California/Oregon samples that formed a single fastStructure population despite covering over 50,000 km2. This result is complemented by the observation that differences among localities represented only 28.87% of the total variation of the northwestern California/Oregon group, which was only slightly higher than the southwestern California group despite their massively different spatial scales and the presence of relatively deep genetic structure between California and Oregon samples in the northwestern California/Oregon group. The genetic divergence of the southwestern California samples emphasizes their extreme importance in conservation and management, given that they likely represent the last vestiges of a population cluster that originally spanned stream habitats from Monterey south to Los Angeles County, a distance of over 500 km (Thomson et al. 2016). The low genetic diversity of the southwestern California samples further points to recent reductions in population size, and emphasizes the challenge they pose for management.
Two key management recommendations stem from this work. First, sequencing of formalin-preserved animals collected from this entire block of extirpated R. boylii populations is becoming increasingly tractable and should be undertaken to help guide future captive breeding and repatriation efforts (Hykin et al. 2015; Ruane and Austin 2017). Second, assisted migration to enable genetic rescue between existing populations within this clade should help retain the genetic variation that remains, and avoid inbreeding depression and reduced long-term fitness decreases in this critically declining population segment (Frankham et al. 2017).
Data archiving
Raw sequence reads are deposited at NCBI (PRJNA401430). Assemblies and variant calls from pyRAD are available at https://doi.org/10.5281/zenodo.885534.
References
Adams AJ, Kupferberg SJ, Wilber MQ, Pessier AP, Grefsrud M, Bobzien S et al. (2017) Extreme drought, host density, sex, and bullfrogs influence fungal pathogen infection in a declining lotic amphibian. Ecosphere 8:e01740
Bradford DF, Knapp RA, Sparling DW, Nash MS, Stanley KA, Tallent-Halsell NG et al. (2011) Pesticide distributions and population declines of California, USA, alpine frogs, Rana muscosa and Rana sierrae. Environ Toxicol Chem 30:682–691
Bryson RW, Savary WE, Zellmer AJ, Bury RB, McCormack JE (2016) Genomic data reveal ancient microendemism in forest scorpions across the California Floristic Province. Mol Ecol 25:3731–3751
Calsbeek R, Thompson JN, Richardson JE (2003) Patterns of molecular evolution and diversification in a biodiversity hotspot: the California Floristic Province. Mol Ecol 12:1021–1029
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Davidson C (2004) Declining Downwind: amphibian population declines in California and historical pesticide use. Ecol Appl 14:1892–1902
Davidson C, Benard MF, Shaffer HB, Parker JM, O’Leary C, Conlon JM et al. (2007) Effects of chytrid and carbaryl exposure on survival, growth and skin peptide defenses in foothill yellow-legged frogs. Environ Sci Technol 41:1771–1776
Davidson C, Shaffer HB, Jennings MR (2002) Spatial tests of the pesticide drift, habitat destruction, UV-B, and climate-change hypotheses for California amphibian declines. Conserv Biol 16:1588–1601
Eaton DAR (2014) PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30:1844–1849
Ecoclub Amphibian Group, Pope KL, Wengert GM, Foley JE, Ashton DT, Botzler RG (2016) Citizen scientists monitor a deadly fungus threatening amphibian communities in northern coastal California, USA. J Wildl Dis 52:516–523
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Felsenstein J (1989) PHYLIP-phylogeny inference package (version 3.2). Cladistics 5:163–166
Frankham R, Ballou JD, Ralls K, Dudash MR (2017) Genetic management of fragmented animal and plant populations. Oxford University Press, Oxford, United Kingdom.
Fu Y-B (2014) Genetic diversity analysis of highly incomplete SNP genotype data with imputations: an empirical assessment. G3 Genes 4:891–900
Hayes MP, Jennings MR (1986) Decline of ranid frog species in western North America: are bullfrogs (Rana catesbeiana) responsible? J Herpetol 20:490–509
Hoffberg SL, Kieran TJ, Catchen JM, Devault A, Faircloth BC, Mauricio R et al. (2016) RADcap: sequence capture of dual-digest RADseq libraries with identifiable duplicates and reduced missing data. Mol Ecol Resour 16:1264–1278
Houlahan JE, Findlay CS, Schmidt BR, Meyer AH, Kuzmin SL (2000) Quantitative evidence for global amphibian population declines. Nature 404:752–755
Hykin SM, Bi K, McGuire JA (2015) Fixing formalin: a method to recover genomic-scale DNA sequence data from formalin-fixed museum specimens using high-throughput sequencing. PLoS ONE 10:e0141579
IUCN, Conservation International, NatureServe (2008) Rana boylii. IUCN 2017. IUCN red list of threatened species. http://www.iucnredlist.org. Accessed 27 Dec 2017.
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI et al. (2017) The K = 2 conundrum. Mol Ecol 26:3594–3602
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HN (ed) Mammalian protein metabolism. Academic Press, New York, p 21–132.
Kerby JL, Sih A (2015) Effects of carbaryl on species interactions of the foothill yellow legged frog (Rana boylii) and the Pacific treefrog (Pseudacris regilla). Hydrobiologia 746:255–269
Kiesecker JM, Blaustein AR, Belden LK (2001) Complex causes of amphibian population declines. Nature 410:681–684
Kuchta SR, Tan A-M (2006) Lineage diversification on an evolving landscape: phylogeography of the California newt, Taricha torosa (Caudata: Salamandridae). Biol J Linn Soc 89:213–239
Kupferberg SJ, Palen WJ, Lind AJ, Bobzien S, Catenazzi A, Drennan JOE et al. (2012) Effects of flow regimes altered by dams on survival, population declines, and range-wide losses of California river-breeding frogs. Conserv Biol 26:513–524
Lapointe F, Rissler LJ (2005) Congruence, consensus, and the comparative phylogeography of codistributed species in California. Am Nat 166:290–299
Lind AJ, Spinks PQ, Fellers GM, Shaffer HB (2011) Rangewide phylogeography and landscape genetics of the Western US endemic frog Rana boylii (Ranidae): implications for the conservation of frogs and rivers. Conserv Genet 12:269–284
Lind AJ, Welsh Jr HH, Wheeler CA (2016) Foothill yellow-legged frog (Rana boylii) oviposition site choice at multiple spatial scales. J Herpetol 50:263–270
Lind AJ, Welsh Jr HH, Wilson RA (1996) The effects of a dam on breeding habitat and egg survival of the foothill yellow-legged frog (Rana boylii) in Northwestern Calfifornia. Herpetol Rev 27:62–66
McCartney-Melstad E, Gidiş M, & Shaffer HB (2017) Population genomics of the Foothill yellow-legged frog (Rana boylii) and RADseq parameter choice for large-genome organisms. BioRxiv. https://doi.org/10.1101/186635
McCartney-Melstad E, Shaffer HB (2015) Amphibian molecular ecology and how it has informed conservation. Mol Ecol 24:5084–5109
Monsen KJ, Blouin MS (2004) Extreme isolation by distance in a montane frog Rana cascadae. Conserv Genet 5:827–835
Myers EA, Rodríguez‐Robles JA, DeNardo DF, Staub RE, Stropoli A, Ruane S et al. (2013) Multilocus phylogeographic assessment of the California Mountain Kingsnake (Lampropeltis zonata) suggests alternative patterns of diversification for the California Floristic Province. Mol Ecol 22:5418–5429
O’Connell KA, Streicher JW, Smith EN, Fujita MK (2017) Geographical features are the predominant driver of molecular diversification in widely distributed North American whipsnakes. Mol Ecol 26:5729–5751
Peek RA (2011) Landscape genetics of Foothill yellow-legged frogs (Rana boylii) in regulated and unregulated rivers: assessing connectivity and genetic fragmentation. Master’s Thesis. University of San Francisco, San Francisco, CA.
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135
Phuong MA, Bi K, Moritz C (2017) Range instability leads to cytonuclear discordance in a morphologically cryptic ground squirrel species complex. Mol Ecol 26:4743–4755
Pickrell JK, Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8:e1002967
R Core Team (2017) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
Raj A, Stephens M, Pritchard JK (2014) fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197:573–589
Reich D, Thangaraj K, Patterson N, Price AL, Singh L (2009) Reconstructing Indian population history. Nature 461:489–494
Richmond JQ, Backlin AR, Tatarian PJ, Solvesky BG, Fisher RN (2014) Population declines lead to replicate patterns of internal range structure at the tips of the distribution of the California red-legged frog (Rana draytonii). Biol Conserv 172:128–137
Rissler H, Graham M, Wake (2006) Phylogeographic lineages and species comparisons in conservation analyses: a case study of California herpetofauna. Am Nat 167:655
Rogers SD, Peacock MM (2012) The disappearing northern leopard frog (Lithobates pipiens): conservation genetics and implications for remnant populations in western Nevada. Ecol Evol 2:2040–2056
Rosenberg NA, Nordborg M (2002) Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nat Rev Genet 3:380–390
Ruane S, Austin CC (2017) Phylogenomics using formalin-fixed and 100 + year-old intractable natural history specimens. Mol Ecol Resour 17:1003–1008
Ryan ME, Palen WJ, Adams MJ, Rochefort RM (2014) Amphibians in the climate vise: loss and restoration of resilience of montane wetland ecosystems in the western US. Front Ecol Environ 12:232–240
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual (3-volume set). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Santos-Barrera G, Hammerson G, Fellers G (2004) Rana boylii. IUCN red list threat species e.T19175A8847383: 1–10.
Spinks PQ, Thomson RC, Shaffer HB (2010) Nuclear gene phylogeography reveals the historical legacy of an ancient inland sea on lineages of the western pond turtle, Emys marmorata in California. Mol Ecol 19:542–556
Spinks PQ, Thomson RC, Shaffer HB (2014) The advantages of going large: genome-wide SNPs clarify the complex population history and systematics of the threatened western pond turtle. Mol Ecol 23:2228–2241
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues ASL, Fischman DL et al. (2004) Status and trends of amphibian declines and extinctions worldwide. Science 306:1783–1786
Subramanian S (2016) The effects of sample size on population genomic analyses – implications for the tests of neutrality. BMC Genom 17:1–13
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tan AM, Wake DB (1995) MtDNA phylogeography of the California newt, Taricha torosa (Caudata, Salamandridae). Mol Phylogenet Evol 4:383–394
Thomson RC, Wright AN, Shaffer HB (2016) California amphibian and reptile species of special concern. University of California Press, Oakland, California
Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A et al. (2014) XSEDE: accelerating scientific discovery. Comput Sci Eng 16:62–74
US Fish and Wildlife Service (2015) Endangered and threatened wildlife and plants; 90-day findings on 31 petitions. 80 FR 37568: 37568–37579.
USDA-NRCS, USGS, EPA (2016) The Watershed Boundary Dataset (WBD) was created from a variety of sources from each state and aggregated into a standard national layer for use in strategic planning and accountability. Watershed Boundary Dataset for the United States of America. <ftp://rockyftp.cr.usgs.gov/vdelivery/Datasets/Staged/Hydrography/WBD/National/GDB/>.
Vähä J-P, Erkinaro J, Niemelä E, Primmer CR (2007) Life-history and habitat features influence the within-river genetic structure of Atlantic salmon. Mol Ecol 16:2638–2654
Vredenburg VT, Bingham R, Knapp R, Morgan JA, Moritz C, Wake D (2007) Concordant molecular and phenotypic data delineate new taxonomy and conservation priorities for the endangered mountain yellow-legged frog. J Zool 271:361–374
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Whiles MR, Hall RO, Dodds WK, Verburg P, Huryn AD, Pringle CM et al. (2013) Disease-driven amphibian declines alter ecosystem processes in a tropical stream. Ecosystems 16:146–157
Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS (2012) A high-performance computing toolset for relatedness and principal component analysis of SNPdata. Bioinformatics 28:3326–3328
Acknowledgements
We thank Amy Lind, the Museum of Vertebrate Zoology, University of California, Berkeley, and the Department of Herpetology at the California Academy of Sciences for providing samples, Erin Toffelmier and Genevieve Mount for laboratory assistance, and Phil Spinks for conversations regarding analyses. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303, and the Comet cluster at the San Diego Supercomputing Center—an XSEDE resource (Towns et al. 2014), supported by NSF ACI-1548562. EMM and HBS are supported by NSF-DEB 1257648 and grants from the US Fish and Wildlife Service and the US Bureau of Reclamation. MG was supported by a grant from The Scientific and Technological Council of Turkey (TUBITAK).
Author contributions
EMM analyzed the data and wrote the initial draft manuscript, MG collected the data and edited the manuscript, and HBS developed the initial project and edited the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
McCartney-Melstad, E., Gidiş, M. & Shaffer, H.B. Population genomic data reveal extreme geographic subdivision and novel conservation actions for the declining foothill yellow-legged frog. Heredity 121, 112–125 (2018). https://doi.org/10.1038/s41437-018-0097-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0097-7
This article is cited by
-
Conservation of biodiversity in the genomics era
Genome Biology (2018)
