
Abstract
Evolutionary success, as demonstrated by high abundance and a wide geographical range, is related to genetic variation and historical demography. Here we assess how climatic change during the Quaternary influenced the demography and distribution of the Neotropical swamp palm Mauritia flexuosa. Using microsatellite loci and coalescent analyses we examined how demographical dynamics affected genetic diversity, effective population size and connectivity through time and space. Mauritia flexuosa presents significant genetic differentiation between the Amazonian and Cerrado biomes and among different river basins. Amazonian lineages are ancient compared to lineages from the Cerrado, a pattern corroborated using the fossil pollen record, where the species was absent from the Cerrado during the cold and dry periods of the last glacial cycles, then returned during the wet, interglacial phases. Coalescent simulations show that the pattern of observed genetic diversity for M. flexuosa is most likely due to a range retraction during the Last Glacial Maximum, leading to multiple refugia and resulting in high differentiation between Amazonian and Cerrado biomes. Isolation-by-distance and by-environment also shaped the distribution and evolutionary success of M. flexuosa. Our study provides new insights into the historical factors that affected geographical distribution and structure genetic diversity, contributing to long-term evolutionary success.
Similar content being viewed by others


Introduction
Evolutionary success and fitness are often treated as synonyms, where evolutionary success is measured by the average contribution of an individual to the gene pool. Haldane (1924) first described the population genetics related to fitness, but it was the expansion to include time and space that allows for the understanding of evolutionary success on geological timescales (Holt 2003). Following this, the contribution to the gene pool and thus evolutionary success, may be increased through three primary avenues: species radiation, species abundance or population size and geographic range (Jablonski 2008).
Radiations lead to lineages with a high number of species, which ensures evolutionary success (Schluter 2000). Fascinating examples of evolutionary radiations abound in the literature, for instance the formation of the African rift lakes has led to the evolution of over 800 cichlid fish species (Joyce et al. 2005), and colonisation of the Andes mountains by Lupinus spurred 81 species (Hughes and Eastwood 2006) to fill geographic space along the cordilleras. Rift lakes and Andean peaks have primarily contributed to species radiation only over the last ca. 2 Ma, since the onset of their current conditions. In contrast, evolutionary success may also be measured by species enduring over a long geological time period, particularly if they have high local abundance. Horseshoe crabs have remained largely morphologically unchanged and abundant for 150 Ma (Avise et al. 1994), similarly for ammonites, yet on a much larger time scale (400 Ma; Knoll and Nowak 2017). Some groups are described as highly successful due to the wide geographical distribution and the variety of environmental conditions they inhabit, such as the African Tilapia (Fryer and Iles 1969).
Evolutionary success has been explored in diverse biogeographical regions, and a landmark study in the most species-rich of all, Amazonia (Myers et al. 2000), has shown that few species dominate Amazonian landscapes (ter Steege et al. 2013). These hyperdominant species consist of a few lineages across the angiosperm tree of life, but the bulk of them are in the palm family (Arecaceae; ter Steege et al. 2013). Among them, Mauritia flexuosa is one of the most common species in Amazonia (Rull 1998; Rull and Montoya 2014), and is likely to be one of the most highly distributed at a global scale (Dransfield et al. 2008), meaning that it is not only highly abundant but has a large geographic range across different biomes, like in Amazonia (Brazil, Colombia, Ecuador, French Guiana, Guyana, Peru, Suriname and Venezuela), in the Llanos (Colombia and Venezuela), and in the Cerrado, the savanna of Central Brazil (Fig. 1). How a species reaches such level of evolutionary success and what factors have contributed to it are still unanswered questions.

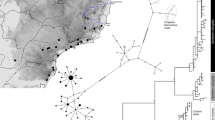
Geographical distribution of the 46 Mauritia flexuosa populations sampled and Bayesian clustering of individuals. Different colours were assigned to different clusters following the figure legends. The lower bar plot (all populations) shows the two clusters (K = 2) when Bayesian analysis was performed considering the 46 populations. The upper bar shows the three clusters for the Amazonia biome (K = 3) and the two clusters for the Cerrado biome (K = 2). For population codes see Supporting Information Table S1. The inset map shows the geographic distribution of M. flexuosa based on the occurrence records (black dots) of GBIF (https://www.gbif.org). The shape file of river basins was obtained from HydroBASINS v. 1c (Lehner and Grill 2013)
Evolutionary success demonstrated by high abundance and a wide geographic range is related to genetic variation and historical demography (Eckert et al. 2008; Parmesan 2006). Stable environments (e.g. the museum model of diversification; Wallace 1878; Fisher 1960) or resilience in the face of climate change can also ensure evolutionary success. Stable environments may have higher carrying capacities and thus support larger effective population sizes (Excoffier et al. 2009; Whitlock and Barton 1997). Widely distributed species that have undergone demographic and range expansion through time, or maintained a wide historical refugium through glacial cycles, have high effective populations sizes, high genetic diversity and historical connectivity (e.g. Collevatti et al. 2012, 2014, 2015a; Vitorino et al. 2016).
During the Last Glacial Maximum (LGM) the climate was generally drier in most of South America, leading to the range retraction of many savanna taxa and the expansion of grasslands in southeast Brazil (Behling 2003; Salgado-Laboriau et al. 1997). However, climate change has driven a diversity of responses across different vegetation types (see Mayle et al. 2004; Van Der Hammen and Hooghiemstra 2000). Western Amazonia was wet during glacial times and dominated by tropical rain forest (e.g. Colinvaux and De Oliveira 2000; Colinvaux et al. 1996), whereas sites in eastern Amazonia were drier and replaced by savanna vegetation during glacial periods (e.g. van der Hammen and Absy 1994). Because glaciations were recurrent throughout the Quaternary, the cycles of range retraction and expansion during glacial and interglacial phases may have caused displacements in species distributions and left genetic signatures (Davis and Shaw 2001). Range expansion, for instance, may lead to a spatial pattern in genome assortment, decreasing genetic diversity in new areas due to leading-edge colonisation (Hewitt 1996). Allele surfing, i.e. the spread and increase of a low-frequency allele or new mutations that migrate on the wave of advance of an expanding population, may also decrease genetic diversity (Arenas et al. 2012; Excoffier and Ray 2008). In addition, due to rapid colonisation and founder effect, density-dependent processes may cause patchy spatial distributions and structuring of genetic diversity, which also decreases genetic diversity in new areas (Excoffier et al. 2009; Waters et al. 2013). Thus, palaeodistribution dynamics may be a key contributor to evolutionary success, affecting geographic range, effective population size and thus long-term population persistence.
Mauritia flexuosa was highly affected by climatic change during the Quaternary in terms of both the species distribution and genetic diversity. Using non-coding chloroplast regions (a single coalescent unit), Lima et al. (2014) found that the geographic range became restricted during glacial times into refugia in the Brazilian savanna. Departing from these results, which were based on populations from only the southeastern portion of the species range (Lima et al. 2014), a synthesis by Rull (1998) suggested that the northern and eastern portions of the M. flexuosa range have been subject to more geological and environmental change than the rest. Species resilience is therefore an important component of the evolutionary success of M. flexuosa as it has persisted throughout drastic changes in climate resulting from glacial cycling, and robust sampling is key for detecting these patterns.
Here we address the role of demographical history in evolutionary success, i.e. high abundance and broad geographic range, studying the hyperdominant species M. flexuosa. Using range-wide sampling, we reconstruct the demographical history of M. flexuosa using coalescent analyses and simulations based on microsatellite loci. The Mauritia lineage originated in Amazonia near the Cretaceous/Tertiary boundary (Rull 1998), and previous work shows a wide historical climatic refugium in Amazonia for M. flexuosa (Lima et al. 2014) that corroborates the history as inferred from pollen fossil records. Moreover, many Cerrado plant lineages evolved from Amazonian lineages (e.g. Simon et al. 2009; Terra-Araújo et al. 2015). Thus, we hypothesise that lineages from Amazonia are older than those from Cerrado and also show high genetic differentiation between the two biomes. Owing to the fact that M. flexuosa inhabits vastly different environmental conditions due to its wide geographical distribution, we also hypothesise that isolation-by-environment (Wang and Bradburd 2014), i.e. a correlation between genetic differentiation among populations and environmental variables, is more important than isolation-by-distance in shaping the genetic differentiation. Lastly, we hypothesise that populations from Amazonia will show high historical connectivity among them, in contrast with Cerrado populations, where pollen fossil records show the disappearance of M. flexuosa during glacial times (see Lima et al. 2014 and references therein). Alternatively, if M. flexuosa persisted in multiple refugia during glaciations, we then expect asymmetrical migration from source populations into sink populations.
Material and methods
Population sampling
We sampled 712 individuals from 46 populations of M. flexuosa throughout its geographic range (Figs. 1 and 2; Table S1). Populations were sampled in Amazonia (24 populations) and the Cerrado (22) biomes across different river basins: 20 populations from the Amazon basin, three in the Caribbean, two in the North Atlantic, three in the Orinoco, 6 in the Paraná-Paraguay, 1 in the São Francisco and 11 in the Araguaia-Tocantins (Fig. 1; Tables S1 and S2). A shape file of river basins was obtained from HydroBASINS v. 1c (Lehner and Grill 2013). Leaves of adults were collected and geographic coordinates were taken for each individual. Because of differences in plant density and population size, the number of individuals sampled differed among localities and ranged from four to 30 (Table S2).

Geographical space of climatic variables in Neotropics during the LGM (21 ka), mid-Holocene (6 ka) and present-day (0 ka), and the geographic distribution of Mauritia flexuosa (black dots). The bioclimatic variables were obtained from AOGCM CCSM4. The black dots are the occurrence records of GBIF (https://www.gbif.org)
Genetic data
Individuals were genotyped using 10 nuclear microsatellite loci (Federman et al. 2012). Primers were marked with fluorescent dyes (6-FAM, HEX and NED, Applied Biosystems, CA) and microsatellite loci amplifications were performed, for each locus separately, in a 10 μL volume containing 0.90 μM of each primer, 1 U Taq DNA polymerase (Phoneutria, BR), 250 μM of each dNTP, 1×reaction buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCL2), 250 μg of BSA and 7.5 ng of template DNA. Amplifications were performed using a PE9700 thermal controller (Applied Biosystems, CA) under the following conditions: 94°C for 5 min (one cycle); 94°C for 1 min, annealing temperature for 1 min, 72°C for 1 min (35 cycles); and 72°C for 30 min (one cycle). The PCR products were subjected to electrophoresis on a GS3500 Genetic Analyzer (Applied Biosystems, CA) and were sized by comparison to a ROX 500 internal lane standard (Applied Biosystems, CA) using Geneious v. 6.1.6 software (Kearse et al. 2012). We used the Micro-Checker software to detect potential errors due to stutter bands, allele dropout and null alleles (Van Oosterhout et al. 2004), and found evidence of null alleles only for a few loci in a few populations (Table S3), which may also be do to inbreeding. We also tested for null alleles using the software INEST (Chybicki and Burczyk 2009) and for most populations we found no difference in inbreeding coefficient estimated with raw data and accounting for null alleles (Table S4). Only two populations, ROY and PNP, showed higher inbreeding when inbreeding was corrected for null alleles (Table S4).
Demography and historical connectivity
Demographic parameters and population structure
We estimated the genetic structure of modern populations of M. flexuosa and determined the number of demes to calibrate coalescent simulations and test the hypothesis of high differentiation between lineages from Amazonia and the Cerrado biomes. We used Bayesian clustering simulations to assess the number of discrete genetic clusters (K) using the software Structure v. 2.3.4 (Pritchard et al. 2000). To minimise the effect of the starting configuration, simulations were performed with a burn-in period of 100,000 repetitions, followed by 1,000,000 Markov-Chain Monte Carlo (MCMC) repetitions of data collection. Four independent runs for each K were performed to evaluate consistency of the results using the admixture model of ancestry and correlated allele frequencies. We used the ΔK method (Evanno et al. 2005) implemented in Structure Harvester v. 0.6.94 (Earl and von Holdt 2012) to detect the number of K that best fits the data.
We performed hierarchical AMOVA implemented in Arlequin v. 3.11 (Excoffier et al. 2005) to test the differentiation between biomes, among river basins and among distinct populations as determined by Structure (see above), partitioning the total variance among groups (FCT), among populations within groups (FSC), among individuals in the total population (FIT), and within individuals (FIS). Statistical significance was tested with a non-parametric test using 10,000 random permutations. Slatkin’s RST (Slatkin 1995) was estimated from the variance in allele sizes to verify the contribution of stepwise-like mutations to genetic differentiation and test the hypothesis that FST = RST based on Hardy et al. (2003) using the software SPAGeDi v. 1.4 (Hardy and Vekemans 2002).
We then performed coalescent analyses (Kingman 1982) implemented in the software Lamarc v. 2.1.8 (Kuhner 2006) to obtain demographic parameters for M. flexuosa. We performed the analyses over all populations to obtain the coalescence time of all M. flexuosa lineages, for each biome and for each river basin, based on results of structure and hierarchical AMOVA (see results below). We estimated theta (θ = 4μNe; coalescent or mutation parameter for a diploid genome, where Ne is effective population size) using an MCMC approach (Beerli and Felsenstein 2001), to estimate coalescence time. To assess historical genetic connectivity, we estimated the number of migrants per generation from a scaled migration rate, M = 4Nem/θ, where m is migration rate. The analyses were run with 20 initial chains of 10,000 steps and three final chains of 100,000 steps that were sampled every 100 steps following a burn-in of 10,000 steps. The analyses were performed four times under the stepwise mutation model and convergence was verified using Tracer v. 1.5 (Drummond and Rambaut 2007) and combined using Log Combiner v. 1.7.1 (Drummond and Rambaut 2007). The results were considered reliable only when effective sample size ≥200.
Time to most recent common ancestor (TMRCA) and effective population size were estimated from the mutation parameter θ (Kingman 1982, see also Hein et al. 2005) using a generation time of 40 years. Generation time was estimated using 12 years as the average time to the first flowering based on data of permanent plots for demography analysis (RG Collevatti, unpublished data) and 50–70 years as a typical life span for palms and that reproductive success is independent of age (Dransfield et al. 2008). We used the microsatellite mutation rate reported for a closely related lineage, Zea mays (Poaceae), of 7.7 × 10−4 (95% CI = 5.2 × 10−4 to 1.1 × 10−3) mutations per allele per generation (Vigouroux et al. 2002). We also chose this fairly low microsatellite mutation rate that generally ranges from 10−2 to 10−4 mutations per allele per generation (Udupa and Baun 2001; Thuillet et al. 2002; Marriage et al. 2009), based on non-significant difference between FST and RST (see results below).
Finally, we obtained parameters of population diversity to compare genetic diversity between biomes and among river basins and to further compare with predicted genetic signatures from alternative demographic hypotheses (see below). We calculated the number of alleles per locus (A) and allelic richness (Ar) based on rarefaction analysis (El Mousadik and Petit 1996) for a reference sample size of five individuals (excluding populations with less than five individuals). Genetic diversity was estimated using the expected heterozygosity (He) under Hardy–Weinberg equilibrium (Nei 1978). The observed heterozygosity (Ho) and the inbreeding coefficient (f) were estimated as a test of deviation from Hardy–Weinberg equilibrium using randomisation-based tests with 10,000 permutations. We then tested whether genetic diversity differs between the Amazonian and Cerrado biomes and among river basins using permutation tests. All analyses were performed with FSTAT v. 2.9.3.2 software (Goudet 2002).
Simulation of demographical history
The demographical history of M. flexuosa was modelled and simulated following the framework described in Collevatti et al. (2012, 2013, 2015b). We simulated six demographical scenarios (Fig. 3). Two scenarios, “Range Expansion” (smaller range size at the LGM compared to present-day) and “Range Stability” (similar range size at the LGM and present-day) were derived from the hypotheses supported by ecological niche modelling (Lima et al. 2014) and one, ‘Multiple Refugia’, was derived from results and interpretation from Ab’Sáber (2000). “Multiple Refugia” confers a retraction of savanna-like vegetation during glacial periods with a highly heterogeneous response of the vegetation, leading to many refugia of different effective sizes, and was the most likely scenario for plastid data in M. flexuosa (Lima et al. 2014). Because of the admixture among populations from the southeastern Amazon basin with populations from Araguaia-Tocantins basin in the Cerrado biome shown by Structure (see results below), we also built demographical scenarios accounting for admixture among populations testing the hypothesis of admixture among populations from the southeastern Amazon basin and from Araguaia-Tocantins river basins after the last glaciation (Fig. 3).

Demographical scenarios simulated for Mauritia flexuosa using the software DIYABC 2.1.0 and their geographic representation. The simulations were performed for six demes. Demes 1, 2 and 3 represent the lineages from the Amazonia biome and demes 4 and 5, and 6 from the Cerrado biome. LGM last glacial maximum, Pres present-day, N0 effective population size at time t0 (present), N1 effective population size at time t1750 (1750 generations ago corresponding to the LGM, 21 ka). The scenarios with admixture test the hypothesis of genetic admixture between populations of the southeastern Amazon basin (deme 6) and populations of Araguaia-Tocantins basin in the Cerrado biome (deme 4) during the LGM
Simulations were performed using the software DIYABC v. 2.1 (Cornuet et al. 2014). For model calibration, we used the mutation rate described above and the demographic parameters estimated using Lamarc. The scenarios were designed using the five clusters recovered by Structure and an additional deme (deme 6, see Fig. 3) comprised of populations from the southeastern Amazon basin (see results below). Population dynamics were simulated backwards from t = 0 (present), with effective size N0 = 3000 for all scenarios, to t = 525 generations ago (at 21 ka), with effective size N1 that shifted among scenarios according to our theoretical expectation (Fig. 3). Given the high variation in M. flexuosa effective population sizes (Table 1), we performed simulations with different initial deme sizes, ranging from N0 = 1000, 3000, 10,000 and 30,000 for all scenarios (around the range of Ne estimated using Lamarc, Table 1). However, we detected differences among scenarios only when N0 = 3000. N0 = 1000 resulted in simulated values (mean number of alleles, allele size and heterozygosity) lower than the observed for M. flexuosa and N0 > 3000 retrieved values higher than the observed.
The mean number of alleles, mean expected heterozygosity, mean allele size variance, mean Gaza-Williamson’s M, and FST were inferred from 600,000 simulations and compared with the observed values and the relative fits of the models were calculated using approximate Bayesian computation (ABC, Excoffier et al. 2005), also implemented in DIYABC. We used the relative proportion of each scenario in the simulated data set closest to the observed data set (hereafter, direct approach) and the logistic regression (hereafter, logistic approach) of each scenario probability on the deviation between simulated and observed summary statistics (Cornuet et al. 2008).
Spatial patterns of genetic diversity
We tested for isolation-by-distance and isolation-by-environment using a multiple regression analysis on matrices of genetic, geographic and environmental distances using a Multiple Matrix Regression with Randomisation approach (MMRR, Wang 2013). For the genetic distance matrix, we used global linearised pairwise FST (Slatkin 1995) and the geographic distance matrix was obtained from the logarithm of the geodesic distances between pair of populations. For environment analysis, we selected the bioclimatic variables from the highest loading on each of the four first factors after performing a factorial analysis with Varimax rotation using the full set of 19 bioclimatic variables (http://www.worldclim.org/bioclim). We selected Bio1 (annual mean temperature), Bio2 (mean diurnal range), Bio14 (precipitation of driest month) and Bio 16 (precipitation of wettest quarter) that explained 86.5% of the variance among populations. We also selected four soil variables from the 30 available from Harmonized World Soil Database (HWSD v 1.2, FAO IIASA ISRIC ISSCAS JRC 2009) using factorial analyses. The variables explained 72.5% of the variance among populations: topsoil density (T_REF_BULK_DENSITY), topsoil organic carbon (T_OC), topsoil clay cationic exchange capacity (T_CEC_CLAY), topsoil salinity (T_ECE). We also performed an autocorrelation analyses using Moran’s I to test for an effect of spatial scale in genetic differentiation implemented in the Spatial Analysis in Macroecology software (SAM, Rangel et al. 2010). Statistical significance of matrix correlations was established from 10,000 random permutations
Because cycles of range retraction during glacial periods and subsequent expansion during interglacial periods may result in a gradient of genetic diversity (Hewitt 1996), we analysed the effect of climate suitability and stability through time on genetic diversity (He) and allelic richness (Ar). For this, we calculated the difference of ensemble suitability among time intervals (i.e. 0–6 ka, 6–21 ka and 0–21 ka) as a measure of climate stability through time using the ecological niche modelling of Lima et al. (2014). We then fit the relationship of climate suitability and stability with both genetic diversity (He) and allelic richness (Ar) using quantile regression (Cade and Noon 2003). The relationship between climate suitability and stability and genetic parameters are usually not linear. Instead, such relationships generally show triangular envelopes surrounding the points, meaning that there are multiple slopes and the central tendency (mean slope) computed by linear methods is not a reliable measure of the relationship (Cade and Noon 2003). In this case, the quantile regression is the suitable method to describe multiple slopes.
We also investigated whether historical changes in geographic range generated a spatial cline in genetic diversity and allelic richness due to expansion, contraction or spatial displacements of climatically suitable conditions. For this, we obtained, for each analysed population, the distance from the centroid of the potential distributions at present-day, 6 ka, and 21 ka, as well as to the centroid of the historical climatic refugium (Lima et al. 2014). We then performed quantile regressions of both genetic diversity (He) and allelic richness (Ar) against these spatial distances from the centroids.
Results
Demography and historical connectivity
Demographic parameters and population structure
The Bayesian simulation grouped populations in two clusters driven by high genetic differentiation between Amazonia and the Cerrado biomes (Fig. 1; Table S5 in Appendix S1 and Figure S1 in Appendix S2). High differentiation between biomes is also seen in the hierarchical AMOVA that showed a high differentiation between biomes (FCT = 0.095, p < 0.001) and among populations within biomes (FSC = 0.137, p < 0.001). On the basis of this result, we performed another structure analysis dividing the biomes and found the Amazonian populations grouped into three clusters and the Cerrado populations clustered into two (Fig. 1, S2, and S3; Tables S6 and S7).
The hierarchical AMOVA showed high genetic differentiation among the three structure clusters in the Amazonian biome (FCT = 0.084, p < 0.001), and similar to the differentiation among populations within clusters (FSC = 0.085, p < 0.001). Differentiation among individuals in the total population was also high (FIT = 0.165, p < 0.001) and inbreeding was not significant (FIS = 0.005, p = 0.363), as expected for a dioecious species. Results were similar in the Cerrado with high and significant differentiation between the two structure clusters (FCT = 0.087, p < 0.001), among populations within clusters (FSC = 0.094, p < 0.001) and among individuals in the total population (FIT = 0.192, p < 0.001). Inbreeding was significant but low (FIS = 0.022, p = 0.008).
Genetic differentiation among river basins was significant (FCT = 0.109, p < 0.001), and differentiation among populations within river basins (FSC = 0.099, p < 0.001) and among individuals in total population (FIT = 0.210, p < 0.001) were also high. The analysis based on allele size also showed high level of genetic differentiation (RST = 0.248, SE = 0.023, p < 0.0001), but not significantly (p = 0.102) different from global FST = 0.186 (SE = 0.028, p < 0.0001), suggesting that stepwise mutations did not cause a shift in mean allele sizes between M. flexuosa populations. We also tested the difference between the two main Structure clusters (Amazonia and Cerrado) for RST = 0.118 (SE = 0.063, p < 0.0001) and FST = 0.109 (SE = 0.027, p < 0.0001), and found no significant difference (p = 0.303).
Populations within river basins had low θ (mutation parameter) and effective population sizes (Table 1), in contrast to high value results for populations when divided amongst Amazonian and the Cerrado biomes. Using the mutation parameter over all populations (θ = 9.021), the TMRCA (Table S8) dated from c. 468.61 ka [95% CI = (371.5–519.5 ka)]. The TMRCA for lineages from Amazonia was more ancient (θ = 4.825), c. 250.65 ka [95% CI = (43.9–519.5 ka)], than for the Cerrado (θ = 2.526) c. 131.21 ka [95% CI = (3.5–464.2 ka)]. Lineages from North Atlantic river basin (c. 877 years) and São Francisco (c. 732 years) were the most recent (see Table S8).
We found high but asymmetrical migration between Amazonia and the Cerrado biomes. The migration from Amazonia into the Cerrado, 10.79 migrants per generation (95% Cl = 0.001–196.363), was lower than from the Cerrado into Amazonia, 18.91 (95% Cl = 0.001–158.243). Adjacent river basins also had high gene flow (Nem ≥ 1.00; Table S9), such as the Amazon, Araguaia-Tocantins and Paraná-Paraguay river basins indicating high historical genetic connectivity. However, historical connectivity was not higher among populations from Amazonia than from the Cerrado (Table S9).
Most populations showed high numbers of alleles per locus and high genetic diversity (He), ranging from 0.522 to 0.885, and non-significant inbreeding (Table S2). Sample size affected the number of alleles sampled in populations because allelic richness (Ar) was lower than the mean number of alleles (A, Table S2). Genetic diversity, allelic richness, inbreeding coefficient and genetic differentiation did not differ between Amazon and Cerrado biomes (Tables S2 and S10), but allelic richness and genetic diversity were significantly higher in the Amazon and Araguaia-Tocantins river basins and lower in Caribbean river basin (Table S10).
Simulation of demographical history
Using the direct approach, we found little variation amongst the different demographical scenarios tested (Figure S4, Table S11). However, results from the logistic approach showed that the scenarios of “Multiple Refugia” with or without admixture were the most probable predictor of the observed genetic parameters of M. flexuosa (Fig. 3 and S4).
Spatial patterns in genetic diversity
Most pairs of populations had high pairwise FST (Figure S5). Genetic differentiation amongst pairs of populations (MMRR, full model r2 = 0.339; p = 0.001) was poorly correlated with geographical distance (r2 = 0.001; p = 0.001). Genetic differentiation among populations was significantly correlated with climatic variables such as mean diurnal temperature range (r2 = 0.339, p = 0.007), but not with annual mean temperature, precipitation of driest month, or precipitation of wettest quarter (Table S12). Genetic differentiation was significantly correlated to topsoil density (r2 = 0.339, p = 0.016) and poorly correlated to topsoil organic carbon (r2 = 0.001, p = 0.007). Autocorrelation analysis showed significant relationship between genetic differentiation and geographical distance for populations up to 800 km (p < 0.005; Figure S6).
Genetic diversity (He) and allelic richness (Ar) were negatively related to the distance from the centroid of the current, 6 ka, and 21 ka estimates of the geographical distribution of the species (Fig. 4, S7, and S8), but not correlated with climatic stability through time (Figures S9 and S10). Areas with higher suitability at present-day, 6 ka, or 21 ka tended to have higher He (Figure S11), but Ar was poorly related to suitability (Figure S12). Moreover, genetic diversity and allelic richness decreased with the distance from the centroid of the historical refugium, but were poorly related with the distance from the edge of the historical refugium (Fig. 4, S13 and S14).

Spatial distribution of genetic diversity of Mauritia flexuosa in relation to the historical refugium, i.e. areas climatically suitable throughout the time (in green). a Allelic richness (Ar). b Expected heterozygosity (He). Circumference sizes are proportional to the value of genetic parameter, following the figure legends
Discussion
Our findings based on microsatellite markers show that historical demography has an important role in the evolutionary success of M. flexuosa. Despite the range retraction during the glacial times, M. flexuosa populations persisted in multiple refugia, leading to high effective population size, historical connectivity and genetic diversity. Although other factors may contribute to evolutionary success such as phenotypic plasticity allowing for survival in both wet (Amazonia) and seasonally dry (Cerrado) biomes, or diversifying selection allowing for adaptation to different climates, here we explicitly explored the role of neutral evolution in the evolutionary success of M. flexuosa driven by demographical history.
Different river basins are inferred to have distinct origins. Lineages from Amazonia (demes 1, 2 and 3 in Fig. 3, Table S8) are more ancient than those from Cerrado (demes 4, 5 and 6 in Fig. 3, Table S8). Within Amazonia, lineages from the Amazon and Orinoco river basins were the most ancient, consistent with previous hypotheses regarding the centre of origin of the species (see Rull 1998 and references therein). It is important to note the partition of M. flexuosa into two highly differentiated lineages, from Amazonia and the Cerrado biomes. The differentiation between biomes seems to be more important than among river basin because populations from the southeastern Amazon basin were clustered with populations from Cerrado biome (CER, COM, VIL, NLA, RIC, POL; Fig. 1; Table S5) and not with other populations of Amazon basin. Our results suggest that the colonisation of the North Atlantic was very recent and occurred from populations of the Caribbean basin towards North Atlantic, which is supported by the more ancient TMRCA of Caribbean lineages (Fig. 1 and Table S8) and the asymmetrical migration (Table S9). The population from the Araguaia-Tocantins river basin (GBE) was most likely derived from North Atlantic lineages. In the Cerrado biome, lineages from the São Francisco river basin were more recent and were derived from Paraná-Paraguay or Araguaia-Tocantins lineages (Fig. 1). In fact, the pollen fossil record shows that the current climatic and vegetation conditions were established only after 4800 year BP in some regions of São Francisco basin (De Oliveira et al. 1999), supporting a recent colonisation of the region.
A demographical scenario of multiple refugia during the LGM was supported from coalescent simulations. Ecological niche modelling and the pollen fossil record show a range shift in the M. flexuosa distribution towards the Amazon basin during the LGM (Lima et al. 2014), where suitable habitats were more available (Fig. 2). Although most of Amazonia comprised a historical refugium for M. flexuosa, the eastern Cerrado was an instable area throughout glacial cycling (Lima et al. 2014). Populations may have undergone extinction in the Cerrado due to the colder and drier conditions of the LGM, or dispersed to and remained constrained within refugia. These refugia may have then served as a source of migrants to other localities, leading to high admixture among populations and the younger TMRCA of lineages from the Cerrado biome.
Although population extinction and subsequent colonisation may also have occurred in Amazonia, the effect was most likely weak due to higher suitability during the LGM and stability through time than in the Cerrado biome. In fact, some sites in southeastern Amazonia were drier during the last glaciation and a grass-dominated savanna without canopy cover replaced the rain forest (Van der Hammen and Hooghiemstra 2000). This scenario reinforces the hypothesis of multiple refugia, even in Amazonia where some sites became drier and may have been sink for migrants in interglacial periods. In addition, admixture of lineages may have occurred numerous times during glacial cycles, as seen by lineage sharing and common ancestry of the southeastern Amazon basin populations and the Cerrado biome even in the chloroplast genome, characterised by its low evolutionary rate and ancient TMRCA (Lima et al. 2014). Multiple refugia during the LGM were also recovered using chloroplast sequences and the pollen fossil record (Lima et al. 2014), corroborating our results on demographical dynamics.
We acknowledge the limitations of modelling demographical scenarios because of the arbitrary assumptions of the priors for model simulation. Although priors are based on demographic parameters estimated from coalescence analyses in Lamarc, the changes in effective population size due to expansion or retraction are based on the range shift predicted by each palaeodistribution model. Moreover, Lamarc software assumes panmixia and the Bayesian genetic clusters are, in fact, a group of local populations that can deviate from panmixia, potentially biasing demographic parameter estimation. To address this issue, we randomly sampled one individual per population and ran the coalescent analyses in Lamarc and DIYABC to check for consistent results. Although effective population sizes (Ne) within river basins were higher than the analysis considering all individuals (Table S13 in Appendix S1), Ne and TMRCA for biomes were slightly smaller, but overall Ne and TMRCA were similar. In addition, coalescent simulation recovered the same scenario of multiple refugia (Figure S15 in Appendix S2). In addition, there is no known mutation rate for M. flexuosa microsatellites and the mutation rate used from a phylogenetically related species may introduce bias to the TMRCA. Despite this, the modelled scenarios based on structure clusters that included populations with higher admixture likely capture the structure, so the potential impact of gene flow among populations in DIYABC estimates was minimised.
The more ancient lineage diversification, higher suitability and climatic stability in Amazonia, and the fact that Amazonia is the centre of origin of M. flexuosa (Rull 1998), may explain the higher diversity and allelic richness in populations of the Amazon basin. This area was also likely closer to the centre of the historical refugium. In fact, western Amazonia remained wetter than the eastern Amazon during glacial times due to Andean influence on climate (Van der Hammen and Hooghiemstra 2000; Hoorn et al. 2010). Populations in the southeastern Amazon basin that remained in the large climatic refugium during glacial times (e.g. CER, VIL, COM, NLA, POL) may have been sources of propagules for subsequent colonisation into the Cerrado, leading to high genetic admixture between these Amazon basin populations with those in the Cerrado. The hypothesis of Cerrado colonisation by Amazonian lineages after glaciation is supported by the ancient age of Amazonian lineages compared to Cerrado, and the pollen fossil records that show complete absence of M. flexuosa in part of the Cerrado biome during the last glaciation (e.g. Behling 2003; Behling and Hooghiemstra 2001; Ledru et al. 1996; Salgado-Labouriau et al. 1998). The establishment of a warmer and wetter climate after the last glaciations may have allowed the colonisation of the Cerrado biome from Amazonia and from local microrefugia in the Cerrado biome.
Our results also show a strong geographic pattern in the distribution of clusters (Fig. 1) that is highly related to biome, river basin, to isolation-by-distance, particularly at shorter distances (<800 km), and to isolation-by-environment. This geographical structure was also recovered by a hierarchical AMOVA that showed high genetic differentiation among populations from Amazonia and the Cerrado biomes and high differentiation among river basins. Mauritia flexuosa relies on watercourses and seeds are adapted to wet environments and are dispersed by water and mammals (Silva et al. 2014), which favours high differentiation among river basins. Isolation-by-environment is mainly due to soil and temperature range variation among populations. Owing to its wide geographical distribution, M. flexuosa faces different environmental conditions in wet (Amazonian rain forest) and seasonally dry (Cerrado) biomes (see Fig. 2) and in different river basins with different geologies, that may lead to genetic differentiation due to natural selection or biased dispersal (Wang and Bradburd 2014).
Anthropogenic effects could bias measurement of evolutionary success, in that any form of domestication alters levels of genetic diversity (Meyer and Purugganan 2013). More importantly, it has recently been suggested that M. flexuosa is hyperdominant because it was domesticated (Levis et al. 2017). Under domestication we expect to find lower genetic diversity in domesticated than in wild populations (Tanksley 1997). In fact, populations sampled in this work show high genetic diversity and number of alleles. Even populations with small sample sizes show a relatively high mean number of alleles per locus and high genetic diversity (e.g. NLA, Table S2, n = 7, A = 6.8, He = 0.839). We compared indices of genetic diversity between populations sampled close to and far from archaeological sites in Amazonia and found no significant differences (see Table S14). Despite the suggestion that M. flexuosa is a domesticated species (Rull and Montoya 2014; Levis et al. 2017), we argue that there is no genetic evidence for domestication in neutral markers. Mauritia flexuosa is associated to watercourses that are often desirable for human settlements, which may explain the association of M. flexuosa and archaeological sites. Humans usually harvest young leaves to make handicrafts and seeds for oil extraction and homemade candy recipes, however these local harvesting practices do not affect the survival, growth, or leaf production of M. flexuosa (Sampaio et al. 2008). Seed dormancy due to physiological and morphological adaptations to swampy environment and recalcitrance (Silva et al. 2014) may actually hamper the domestication of this palm species as seeds may not be easily stored and germinated.
Although Amazonia and the Cerrado biomes have similar genetic diversity, the Amazon and Araguaia-Tocantins river basins had higher genetic diversity compared to the other river basins. Most populations in the Amazon and Araguaia-Tocantins basins are inside the historical climatic refugium and are in climatically suitable sites through geological time (Fig. 4). This stability through time may have allowed for species persistence and higher effective populations sizes, increasing genetic diversity. The Amazon and Araguaia-Tocantins river basins also had high historical connectivity, which may explain their higher genetic diversity. Other palm species show lower genetic diversity compared to M. flexuosa, such as Livistona carinensis (Shapcott et al. 2009), Carpoxylon macrospermum (Dowe et al. 1997), Ptychosperma macarthurii (Shapcott 1998). Those palm species have smaller populations compared to M. flexuosa, which may cause the lower genetic diversity (Shapcott et al. 2009). On the other hand, Euterpe edulis (Carvalho et al. 2017), a widely distributed Neotropical palm species show similar genetic diversity compared to M. flexuosa. Domesticated palm species such as Elaeis guineensis (He = 0.644; Bakoumé et al. 2015) and Cocos nucifera (He ranging from 0.40 to 0.62; Loiola et al. 2016) have lower genetic diversity than M. flexuosa.
In conclusion, our findings show high differentiation among populations of M. flexuosa from Amazonian and the Cerrado biomes. Amazonian lineages are older than those from the Cerrado. The genetic admixture among populations of the southeastern Amazon and Araguaia-Tocantins river basins suggests a colonisation of the western Cerrado by lineages from Amazonia, which is also supported by asymmetrical migration from the Amazon basin into the Araguaia-Tocantins and a more ancient TMRCA. Using microsatellite markers we recovered the demographical history over the last c. 468.6 ka showing that the demographical dynamics of multiple refugia during the LGM, together with the soil and climatic environmental differences between biomes and river basins likely favoured the evolutionary success of M. flexuosa.
Data archiving
Additional data are provided as supporting information in the online version of this article.
References
Ab’Sáber AN (2000) The natural organization of Brazilian inter- and subtropical landscapes. Rev Do Inst Geológico 21(1/2):57–70
Arenas M, Ray N, Currat M, Excoffier L (2012) Consequences of range contractions and range shifts on molecular diversity. Mol Biol Evol 29:207–218
Avise JC, Nelson WS, Sugita H (1994) A speciational history of living fossils—molecular evolutionary patterns in horseshoe crabs. Evolution 48:1986–2001
Bakoumé C, Wickneswari R, Siju S, Rajanaidu N, Kushairi A, Billotte N (2015) Genetic diversity of the world’s largest oil palm (Elaeis guineensis Jacq.) field genebank accessions using microsatellite markers. Genet Resour Crop Evol 62:349–360
Beerli P, Felsenstein J (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA 98:4563–4568
Behling H (2003) Late glacial and Holocene vegetation, climate and fire history inferred from Lagoa Nova in the southeastern Brazilian lowland. Veg Hist Archaeobotany 12:263–270
Behling H, Hooghiemstra H (2001) Chap. 18—Neotropical Savanna environments in space and time: Late Quaternary Interhemispheric ComparisonsA2–Markgraf, Vera BT–interhemispheric climate linkages. Academic Press, San Diego, pp 307–323
Cade BS, Noon BR (2003) A gentle introduction to quantile regression for ecologists. Front Ecol Environ 1:412–420
Carvalho CS, Ballesteros-Mejia L, Ribeiro MC, Côrtes MC, Santos AS, Collevatti RG (2017) Climatic stability and contemporary human impacts affect the genetic diversity and conservation status of a tropical palm in the Atlantic Forest of Brazil. Conserv Genet 18:467–478
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Colinvaux PA, De Oliveira PE (2000) Palaeoecology and climate of the Amazon basin during the last glacial cycle. J Quat Sci 15:347–356
Colinvaux PA, De Oliveira PE, Moreno JE, Miller MC, Bush MB (1996) A long pollen record from lowland Amazonia: forest and cooling in glacial times. Science 274:85–88
Collevatti RG, Lima-Ribeiro MS, Terribile LC, Guedes LBS, Rosa FF, Telles MPC (2014) Recovering species demographic history from multi-model inference: the case of a Neotropical savanna tree species. BMC Evol Biol 14:213
Collevatti RG, Terribile LC, Oliveira G, Lima-Ribeiro MS, Nabout JC, Rangel TF, Diniz-Filho JAF (2013) Drawbacks to palaeodistribution modelling: the case of South American seasonally dry forests. J Biogeogr 40:345–358
Collevatti RG, Terribile LC, Lima-Ribeiro MS, Nabout JC, Oliveira G, Rangel TF, Rabelo SE, Diniz-Filho JAF (2012) A coupled phylogeographical and species distribution modelling approach recovers the demographical history of a Neotropical seasonally dry forest tree species. Mol Ecol 21:5845–5863
Collevatti RG, Terribile LC, Rabelo SG, Lima-Ribeiro MS (2015a) Relaxed random walk model coupled with ecological niche modeling unravel the dispersal dynamics of a Neotropical savanna tree species in the deeper Quaternary. Front Plant Sci 6:1–15
Collevatti RG, Terribile LC, Diniz-Filho JAF, Lima-Ribeiro MS (2015b) Multi-model inference in comparative phylogeography: an integrative approach based on multiple lines of evidence. Front Genet 5:1–8
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R et al. (2014) DIYABCv2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30:1187–1189
Cornuet JM, Santos F, Beaumont MA, Robert CP, Marin JM, Balding DJ et al. (2008) Inferring population history with DIYABC: a user-friendly approach to approximate Bayesian computation. Bioinformatics 24:2713–2719
Davis MB, Shaw RG (2001) Range shifts and adaptive responses to quaternary climate change. Science 292:673–679
De Oliveira PE, Barreto AMF, Suguio K (1999) Late Pleistocene/Holocene climatic and vegetational history of the Brazilian caatinga: the fossil dunes of the middle Sao Francisco River. Palaeogeogr Palaeoclimatol Palaeoecol 152:319–337
Dowe JL, Benzie J, Ballment E (1997) Ecology and genetics of Carpoxylon macrospermum H. Wendl. and Drude (Arecaceae), an endangered palm from Vanuatu. Biol Conserv 79:205–216
Dransfield J, Uhl NW, Lange C, Baker WJ, Harley M, Lewis C (2008) Genera Palmarum. The evolution and classification of palms. International Palm Society, London
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol 17:1170–1188
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree Argania spinosa (L.) Skeels endemic to Morocco. Theor Appl Genet 92:832–839
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Excoffier L, Ray N (2008) Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol 23:347–351
FAO, IIASA, ISRIC, ISSCAS, JRC (2009) Harmonized world soil database (version 1.1). FAO and IIASA, Rome, Italy and Laxenburg, Austria
Federman S, Hyseni C, Clement W, Caccone A (2012) Isolation of 13 novel highly polymorphic microsatellite loci for the Amazonian palm Mauritia flexuosa L.f. (Arecaceae). Conserv Genet Resour 4:355–357
Fischer AG (1960) Latitudinal variations in organic diversity. Evolution 14:64–81
Fryer G, Iles TD (1969) Alternative routes to evolutionary success as exhibited by African Cichlid fishes of the genus Tilapia and the species flocks of the Great Lakes. Evolution 23:359–369
Goudet J (2002). FSTAT, a program to estimate and test gene diversities and fixation indices. Version 2.9.3.2. http://www2.unil.ch/popgen/softwares/fstat.htm
Haldane JBS (1924) A mathematical theory of natural and artificial selection. Part II: the influence of partial self-fertilisation, inbreeding, assortative mating, and selective fertilisation on the composition of Mendelian populations, and on natural selection. Biol Rev 1:158–163
Hardy OJ, Charbonnel N, Fréville H, Heuertz M (2003) Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163:1467–1482
Hardy O, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hein J, Schierup MH, Wiuf C (2005) Gene genealogies, variation and evolution: a primer in coalescent theory. Oxford University Press, London
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276
Holt RD (2003) On the evolutionary ecology of species’ ranges. Evolut Ecol Res 5:159–178
Hoorn C, Wesselingh FP, ter Steege H, Bermudez MA, Mora A, Sevink J et al. (2010) Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science 330:927–931
Hughes C, Eastwood R (2006) Island radiation on a continental scale: exceptional rates of plant diversification after uplift of the Andes. Proc Natl Acad Sci 103:10334–10339
Jablonski D (2008) Species selection: theory and data. Annu Rev Ecol Evol Syst 39:501–524
Joyce DA, Lunt DH, Bills R, Turner GF, Katongo C, Duftner N et al. (2005) An extant cichlid fish radiation emerged in an extinct Pleistocene lake. Nature 435:90–95
Kearse M, Moir R, Wilson A, Stones-Havas S, Sturrock S, Buxton S et al. (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kingman JFC (1982) The coalescent. Stoch Process Appl 13:235–248
Knoll AH, Nowak MA (2017) The timetable of evolution. Sci Adv 3:1–13
Kuhner MK (2006) LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 22:768–770
Ledru M, Braga S, Soubies F, Fournier M, Martin L, Suguio K, Turcq B (1996) The last 50,000 years in the Neotropics (Southern Brazil): evolution of vegetation and climate. Palaeogr Palaeoclimatol Palaeoecol 123:239–257
Lehner B, Grill G (2013) Global river hydrography and network routing: baseline data and new approaches to study the world’s large river systems. Hydrol Process 27:2171–2186. www.hydrosheds.org
Levis C, Costa FRC, Bongers F, Peña-Claros M, Clement CR, Junqueira AB et al. (2017) Persistent effects of pre-Columbian plant domestication on Amazonian forest composition. Science 355:925–931
Lima NE, Lima-Ribeiro MS, Tinoco CF, Terribile LC, Collevatti RG (2014) Phylogeography and ecological niche modelling, coupled with the fossil pollen record, unravel the demographic history of a Neotropical swamp palm through the quaternary. J Biogeogr 41:673–686
Loiola CM, Azevedo AON, Diniz LEC, Aragão WM, Carlos CD, Santos PHAD et al. (2016) Genetic relationships among tall coconut palm (Cocos nucifera L.) accessions of the international coconut genebank for Latin America and the Caribbean (ICG-LAC), evaluated using microsatellite markers (SSRs). PLoS ONE 11:1–11
Marriage TN, Hudman S, Mort ME, Orive ME, Shaw RG, Kelly JK (2009) Direct estimation of the mutation rate at dinucleotide microsatellite loci in Arabidopsis thaliana (Brassicaceae). Heredity 103:310–317
Mayle FE, Beerling DJ, Gosling WD, Bush MB (2004) Responses of Amazonian ecosystems to climatic and atmospheric carbon dioxide changes since the last glacial maximum. Philos Trans R Soc Lond B Biol Sci 359:499–514
Meyer RS, Purugganan MD (2013) Evolution of crop species: genetics of domestication and diversification. Nat Rev Genet 14:840–852
Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Parmesan C (2006) Ecological and evolutionary responses to recent climate change. Annu Rev Ecol Evol Syst 37:637–669
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rangel TF, Diniz-Filho JAF, Bini LM (2010) SAM: a comprehensive application for spatial analysis in macroecology. Ecography 33:46–50
Rull V (1998) Biogeographical and evolutionary considerations of Mauritia (Arecaceae), based on palynological evidence. Rev Palaeobot Palynol 100:109–122
Rull V, Montoya E (2014) Mauritia flexuosa palm swamp communities: natural or human-made? A palynological study of the Gran Sabana region (northern South America) within a Neotropical context. Quat Sci Rev 99:17–33
Salgado-Laboriau ML, Casseti V, Ferraz-Vicentini KR, Martin L, Soubiés F, Suguio K, Turcq B (1997) Late quaternary vegetacional and climatic changes in Cerrado and palm swamp from Central Brazil. Palaeogeogr Palaeoclimatol Palaeoecol 128:215–226
Salgado-Labouriau ML, Barberi M, Ferraz-Vicentini KR, Parizzi MG (1998) A dry climatic event during the late quaternary of tropical Brazil. Rev Palaeobot Palynol 99:115–129
Sampaio MB, Schmidt IB, Figueiredo IB (2008) Harvesting effects and population ecology of the Buriti Palm (Mauritia). Econ Bot 62:171–181
Shapcott A (1998) The genetics of Ptychosperma bleeseri, a rare palm from the Northern Territory, Australia. Biol Conserv 85:203–209
Shapcott A, Dowe JL, Ford H (2009) Low genetic diversity and recovery implications of the vulnerable Bankoualé palm Livistona carinensis (Arecaceae), from North-eastern Africa and the Southern Arabian Peninsula. Conserv Genet 10:317–327
Silva RS, Ribeiro LM, Mercadante-Simões MO, Nunes YRF, Lopes PSN (2014) Seed structure and germination in buriti (Mauritia flexuosa), the swamp palm. Flora: Morphol Distrib Funct Ecol Plants 209:674–685
Simon MF, Grether R, de Queiroz LP, Skema C, Pennington RT, Hughes CE (2009) Recent assembly of the Cerrado, a neotropical plant diversity hotspot, by in situ evolution of adaptations to fire. Proc Natl Acad Sci 106:20359–20364
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Schluter D (2000) The ecology of adaptive radiation. Oxford University Press, New York, NY, p 296
Tanksley SD (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
ter Steege H, Pitman NCA, Sabatier D, Baraloto C, Salomao RP, Guevara JE et al. (2013) Hyperdominance in the Amazonian tree flora. Science 342:1243092–1243092
Terra-Araujo MH, de Faria AD, Vicentini A, Nylinder S, Swenson U (2015) Species tree phylogeny and biogeography of the Neotropical genus Pradosia (Sapotaceae, Chrysophylloideae). Mol Phylogenet Evol 87:1–13
Thuillet AC, Bru D, David J, Roumet P, Santoni S, Sourdille P et al. (2002) Direct estimation of mutation rate for 10 microsatellite loci in durum wheat, Triticum turgidum (L.) Thell. ssp durum desf. Mol Biol Evol 19:122–125
Udupa S, Baum M (2001) High mutation rate and mutational bias at (TAA)n microsatellite loci in chickpea (Cicer arietinum L.). Mol Genet Genom 265:1097–1103
Van der Hammen T, Absy ML (1994) Amazonia during the last glacial. Palaeogeogr Palaeoclimatol Palaeoecol 109:247–261
Van Der Hammen T, Hooghiemstra H (2000) Neogene and quaternary history of vegetation, climate, and plant diversity in Amazonia. Quat Sci Rev 19:725–742
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vigouroux Y, Jaqueth JS, Matsuoka Y, Smith OS, Beavis WD, Smith JSC, Doebley J (2002) Rate and pattern of mutation at microsatellite loci in maize. Mol Biol Evol 19:1251–60
Vitorino LC, Lima-Ribeiro MS, Terribile LC, Collevatti RG (2016) Demographical history and palaeodistribution modelling show range shift towards Amazon Basin for a Neotropical tree species in the LGM. BMC Evol Biol 16:213
Wallace AR (1878) Tropical nature, and other essays. Macmillan & Co, London
Wang IJ (2013) Examining the full effects of landscape heterogeneity on spatial genetic variation: a multiple matrix regression approach for quantifying geographic and ecological isolation. Evolution 67:3403–3411
Wang IJ, Bradburd GS (2014) Isolation by environment. Mol Ecol 23:5649–5662
Waters JM, Fraser CI, Hewitt GM (2013) Founder takes all: density-dependent processes structure biodiversity. Trends Ecol Evol 28:78–85
Whitlock MC, Barton NH (1997) The effective size of subdivided population. Genetics 146:427–441
Acknowledgements
This work was supported by a grant from the Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq/PVE no. 407310/2013-4) to RGC and CDB. RGC was also supported by the research network Rede Cerrado CNPq/PPBio (Project No. 457406/2012-7) and CAPES/PROCAD (Project No. 88881.068425/2014-01). WM received a FAPEG fellowship and CGF a CNPq fellowship. We thank the Editor, Dr. Barbara Marble, for comments that improved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Melo, W.A., Freitas, C.G., Bacon, C.D. et al. The road to evolutionary success: insights from the demographic history of an Amazonian palm. Heredity 121, 183–195 (2018). https://doi.org/10.1038/s41437-018-0074-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0074-1
This article is cited by
-
Recent demographic histories of temperate deciduous trees inferred from microsatellite markers
BMC Ecology and Evolution (2021)
-
Genetic structure of two Acrocomia ecotypes (Arecaceae) across Brazilian savannas and seasonally dry forests
Tree Genetics & Genomes (2020)
-
Cytological and histochemical evaluations reveal roles of the cotyledonary petiole in the germination and seedling development of Mauritia flexuosa (Arecaceae)
Protoplasma (2019)
