
Abstract
Disease-associated variants in the human genome are continually being identified using DNA sequencing technologies that are especially effective for Mendelian disorders. Here we sequenced whole genome to high coverage (>30×) of 6 members of a 7-generation family with dwarfism from a consanguineous tribe in Pakistan to determine the causal variant(s). We identified a missense variant rs111033552 (c.2011T>C [p.Ser671Pro]) located in COL10A1 (encodes the alpha chain of type X collagen) as the most likely contributor to the dwarfism. We further confirmed the variant in 22 family members using Sanger sequencing. All affected individuals are heterozygous for the missense mutation rs111033552 and no individual homozygous was observed. Moreover, the mutation was absent in 69,985 individuals representing >150 global populations. Taking advantage of whole-genome sequencing data, we also examined other variant forms, including copy number variation and insertion/deletion, but failed to identify such variants enriched in the affected individuals. Thus rs111033552 had priority for linkage with dwarfism.
Similar content being viewed by others

Introduction
Human height is a classical quantitative trait and previous genome-wide association studies have identified hundreds of loci or genes that contribute to height predisposition (Chan et al. 2015; He et al. 2015; Wood et al. 2014). However, the association strategies usually explain a small proportion of the observed phenotypic variance under study, which is considered “missing heritability” (Koch 2014; Manolio et al. 2009). In contrast, in some special cases, height could be a Mendelian trait and strongly affected by a single locus. Skeletal dysplasias are the most common type of dwarfism, characterized by abnormal bone growth and a disproportionate body. A distinct example is metaphyseal chondrodysplasia, Schmid type (MCDS) [OMIM, 156500] which is characterized by short stature with abnormally short arms and legs (short-limbed dwarfism) and bowed legs (genu varum), coxa valga, metaphyseal widening and sclerosis (Ikegawa et al. 1997, 1998; McIntosh et al. 1994; Wallis et al. 1996). Next-generation sequencing (NGS) is a new powerful tool for identifying genes that underlie Mendelian disorders (Bamshad et al. 2011; Ng et al. 2010). We enrolled a 7-generation dwarf family with typical MCDS syndromes from an isolated consanguineous tribe in Pakistan to identify the gene(s) associated with MCDS to determine the causal genetic variant(s). Whole-genome deep sequencing (WGS) was used to offer complete coverage of the coding region of the genome and it allows examination of variants in non-coding regions, without omitting regulatory regions such as promoters and enhancers (Meienberg et al. 2016).
Materials and methods
Blood samples were collected from an isolated consanguineous tribe in the Sikhaniwala region in District Rajanpur of Punjab Province (Pakistan). Written informed consents were obtained from all subjects at the time of enrollment. All experimental protocols and subject handling procedures were approved by the Biomedical Research Ethics Committee of Shanghai Institutes for Biological Sciences. All procedures were in accordance with the ethical standards of the Responsible Committee on Human Experimentation (approved by the Biomedical Research Ethics Committee of Shanghai Institutes for Biological Sciences) and the Helsinki Declaration of 1975, as revised in 2000.
Six individuals (VI-1, VI-5, V-2, V-19, VI-27, and VI-30) were selected for WGS (Fig. 1 and Table 1). WGS was carried out on Illumina HiSeq X Ten, with a target high coverage (30–60×) for 150 bp paired-end reads, following Illumina-provided protocols with standard library preparation in WuXiNextCODE. Each sample was run on a unique lane with at least 90 GB data that passed filtering and the reads data were quality controlled to ensure that 80% of the bases achieved at least a base quality score of 30. Reads were mapped to the human reference genome GRCh37 using Burrows-Wheeler Algorithm (Li and Durbin 2010). Single-nucleotide polymorphism (SNP) calling and raw variants filtering were carried out using the HaplotypeCaller module and the variant quality score recalibration module in GATK (DePristo et al. 2011; McKenna et al. 2010), respectively. Copy number variations (CNVs) were called by CNVnator with bin size set as 100 kb (Abyzov et al. 2011). Genotype quality control followed the pipeline described elsewhere (Lu et al. 2016; Mallick et al. 2016). The summary of the NGS data and the called variants are listed in Tables S1 and S2, respectively.

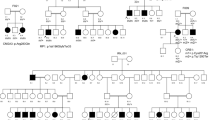
a Pedigree of 7 generations of 110 individuals among which 47 are affected with dwarfism. Dashed line represents the same individuals who are offspring of consanguineous marriage. Red arrows denote individuals with whole genomes sequenced with NGS. b Radiographic features of lower limbs of an MCDS patient at 15 years of age (VI-10 shown in a). c A normal individual (VI-14 shown in a). MCDS patient had bowed lower legs, coxa valga, metaphyseal widening and sclerosis, and radiography of the normal subjects is quite normal.
The overall framework for data analysis is illustrated in Figure S1. To narrow causal variants, we used pedigree information as a first line of filtering. Normal individuals should be homozygous wild type and affected ones should harbor at least one mutated allele (Tables S3 and S4), since the inheritance mode of this disorder is autosomal dominant. Assuming a pathogenic allele to be extremely rare, we then filtered variants against a set of high allele frequency polymorphisms (>0.5%) in worldwide populations according to public databases (dbSNP, the 1000 genomes project (1000 Genomes Project et al. 2015), the NHLBI Exome Sequencing Project (ESP) database and the Exome Aggregation Consortium (ExAC) database (Bahcall 2016) (Table S3). We further annotated and ranked the genetic variants by type according to genetic location, conservation according to combined annotation-dependent depletion (CADD), GERP, Polyphon2, and SIFT, and pathogenicity according to OMIM and ClinVar databases (Table S3). All genetic annotation was performed with a variant effect predictor tool (VEP) (McLaren et al. 2016) and our in-house python script.
A standard Sanger sequencing approach was used to identify genotypes of 18 candidate SNPs screened from NGS data. Primers were designed with Primer 3.0 (Table S4). PCR was performed with HotStarTaq DNA Polymerase (Qiagen Inc.). A 20-µl mixture was prepared for each reaction and included 1 × GC buffer I (TAKARA), 2.5 mg Mg2+, 0.2 mg dNTP, 0.2 µM of each primer, 1 U HotStarTaq DNA Polymerase (Qiagen Inc.) and 1 µl of dilution DNA at a concentration of 10ng/µl, 0.5 U SAP and 4 U Exo I were added into 8 µl PCR product for purification. The mixture was incubated at 37 °C for 60 min, followed by incubation at 75 °C for 15 min. Purified PCR products were sequenced with a Big-Dye Terminator Cycle Sequencing Kit and an ABI 3730XL Genetic Analyzer (Applied Biosystems) and raw data were analyzed with PolyPhred 6.18.
X-ray radiography was performed to verify the symptom of dwarfism with lower limbs for an affected individual VI-10 and a normal individual VI-14 (Fig. 1b).
We applied LOD score, that is, logarithm (base 10) of odds, a statistical test commonly used for linkage analysis, to validate the association between the candidate locus (rs111033552) and MCDS in the family (V-1, V-2, VI-1, VI-3, VI-4, VI-5, VI-6, VI-7, and VI-8) from the pedigree. The LOD score compares the likelihood of obtaining the test data if two loci are indeed linked to the likelihood of observing the same data purely by chance. A LOD score >3.0 is considered evidence for linkage, while a LOD score <−2.0 indicates no linkage.
Results
The 7-generation pedigree from a family with dwarfism consisted of 110 members, including 47 individuals with short stature and 63 individuals with normal heights (Fig. 1a). The trait of short stature was brought to the pedigree by a female from the second generation, II-6, the first person defined as dwarf in the family (Fig. 1a). The average height of the affected individuals and non-affected ones were 128.5 ± 13.1 and 160 ± 14.5 cm, respectively (Table 1). Radiographic features of lower limbs of the affected individual (VI-10) are bowed lower legs, coxa valga, metaphyseal widening, and sclerosis, while those of the normal individual (VI-14) are quite normal. These identifiable features of lower limb presented in affected individuals are in accordance with that of MCDC, an autosomal dominantly inherited cartilage disorder (Bateman et al. 2005; Ikegawa et al. 1998) (Fig. 1b). Intellectual disability was not identified in any enrolled subject.
To identify gene(s) and variant(s) in the MCDS pedigree, we sequenced whole genomes of 6 individuals (VI-1, VI-5, V-2, V-19, VI-27, and VI-30 in Fig. 1a). WGS analysis revealed 5.02 million SNPs including 4.9% novel SNPs (dbSNP147) (Tables S1 and S2). After sequencing and data filtration (Figure S1), we obtained 29,123 variants meeting requirements of mode of dominant inheritance and segregation in families (Table S3). Required variants should be with allele frequencies <0.5% across worldwide populations, so 1370 variants were left for further filtration. We pinpointed the damaging missense single-nucleotide variant (SNV), rs111033552 located in the third exon, at nucleotide 2011of COL10A1 as the most likely causal variant underlying the dwarfism in this isolated family based on pathogenicity and conservation prediction. Each affected sequenced subject carried a heterozygous allele (A/G) at rs111033552, while normal individuals had reference homozygotes (Fig. 2a). Moreover, we calculated LOD score for rs11103355 and obtained a value of 4.16. This result, in conjunction with WGS filtering approaches we used, supported that rs11103355 is likely the causal variant.

a Six individuals from two families sequenced with NGS. According to the mode of inheritance, individuals with normal height (VI-1 and VI-30) carried homozygous alleles (A/A) while affected ones (V-2, VI-5, V-19, and VI-27) harbor heterozygous alleles (A/G) in rs111033552. b The mutation (from A to G) of rs111033552 was confirmed in 22 family members (Table 1) by Sanger sequencing. c Allele frequencies of G in the pedigree and worldwide populations or databases. The G allele is absent in ExAC, the 1000 genomes project database, Simons genome diversity project, and the NHLBI Exome Sequencing Project (ESP) database. However, the allele frequency (AF) of G in affected and unaffected individualsin the pedigree is 1 and 0, respectively. The AF in the pedigree was estimated using Sanger sequencing of 22 individuals as shown in b. A for affected and UA for unaffected. AF in South Asian, East Asian, European, African, and American were obtained from the 1000 genomes project. ESP NHLBI Exome Sequencing Project (ESP) database, ExAC the Exome Aggregation Consortium (ExAC) database.
Further, we validated the variant in 22 individuals (Table 1) by examining COL10A1 region and another 17 candidate genes using Sanger sequencing (Tables S4 and S5). Among the 22 samples, all individuals (17) with short stature harbored a heterozygous mutation (A/G) at rs111033552 (Fig. 2b). The other five normal individuals carried normal genotype at this genomic position. This variant is extremely rare and has never been reported in Exome Aggregation Consortium (ExAC) database, the 1000 genomes project database (1000 Genomes Project et al. 2015), Simons genome diversity project (Mallick et al. 2016), or NHLBI Exome Sequencing Project (ESP) database (Fig. 2c).
Conservation is a critical criterion for measuring the importance of a certain variant on an evolutionary scale. The CADD score, which ranks deleteriousness of SNVs within the human genome, is 17.4 for rs111033552, indicating that it is predicted to be in the top 2.5% of most damaging derangements for the genome. The 671st residue on which rs111033552 is located is extremely conserved from zebrafish to human (GERP++ score of 3.88; Fig. 3) and this variant is predicted to be deleterious according to several prediction algorithms, including Phylop score, phastCons, PolyPhen (probably damaging (0.993)), SIFT (deleterious (0)), and ClinVar (Pathogenic) (Fig. 3). These results support the contention that the amino acid change made by rs111033552 is likely to exert a specific functional effect on the protein.

Results of comparative genomics show exon 3 in COL10A1 is extremely conserved from fishes to mammals sharing a vast majority of the protein sequences. rs111033552 is predicted to be damaging or pathogenic according to SIFT (deleterious (0)), PolyPhen2 (probably damaging (0.993)) as well as ClinVar database. Tracks here are from the UCSC genome browser.
When the variant rs111033552 changes the 671st amino acid (located in the third exon) of COL10A1 from Serine to Proline (p.Ser671Pro), it changes a polar side-chain to non polar side-chain (Figs. 3 and 4). COL10A1 encodes the alpha chain (α1 [X]) of type X collagen, which is a homotrimer of three α1 (X) chains, each comprised of collagenous domain (COL1) flanked by a N-terminal noncollagenous domain (NC2) and C-terminal noncollagenous NC1 domain (Bateman et al. 2005). Of note, the 3-kb terminal exon (exon 3) of COL10A1 codes for most of the protein sequence of collagen X, including the NC1, the entire COL1 domain, and part of the NC2 domain (Bateman et al. 2005). During endochondral ossification, collagen X is highly expressed by hypertrophic chondrocytes, which are key to bone development (Bateman et al. 2005; Schmid and Linsenmayer 1985). Failed assembly of the homotrimer of collagen X can cause severe bone disorders.

NC1 is encoded by exon 3 of COL10A1. Blue arrows show the position of the missense mutation (rs111033552) that changes the 671st amino acid of COL10A1 from Serine to Proline (p.Ser671Pro). Structure obtained from NCBI structure database.
Discussion
In addition to rs111033552, which was first identified in an US family with dwarfism in 1996 (Stratakis et al. 1996), approximately 50 mutations in COL10A1 have been reported in families of different ethnic origins with MCDS across the globe (Bateman et al. 2005; Higuchi et al. 2016). The variant rs111033552 might play a role in preventing the formation of stable NC1 trimers collagen X, as it is located within the solvent-filled region of the trimer center and changes the amino acid polarity (Bateman et al. 2005) (Fig. 4). Based on the collected pedigree, the mutation is heterozygous and the homozygous form (G/G) is lethal. An individual with A/G genotype of rs111033552 is not able to express sufficient protein needed for conformation of collagen X homotrimer and this would lead to MCDS. Single base changes in COL10A1 have been divided into three classes: class I mutations influence residues within the hydrophobic core of the NC1 domain and were predicted to cause misfolding of NC1 domain; class II mutations are located at the surface of the monomer that is associated with intersubunit assembly were predicted to prevent normal trimer formation; class III mutations affect residues located at the surface of trimer and were predicted to affect interactions associated with the supramolecular assembly of collagen X. Moreover, nonsense and frameshift mutations in COL10A1 were also studied in MCDS patients. Nonetheless, few mutations have been validated for their pathogenic mechanism in vitro and in vivo (Ho et al. 2007), leaving the function of the major variants unresolved. Advances in conservation prediction algorithms and the abundance of human genomic data enable us to estimate the functional importance of these variants with unknown function. We therefore prioritize variants associated with MCDS including rs111033552 and those collected from previous studies (Table S6). Our analysis showed that this mutation is one of the 12 variants absent from global populations and is highly conserved, indicating that the genetic basis of MCDS is highly heterogeneous and such allelic heterogeneity is pervasive in other Mendelian disorders (Pritchard and Cox 2002).
The known history of miscarriages and stillbirths in the family suggested that homozygous mutated genotype (G/G) of rs111033552 is lethal. For example, IV-13 and VI-3 both had one abortion with developmental abnormalities. In both cases, the couples carried a heterozygous genotype, that is, A/G, thus the pregnancy loss was likely due to a homozygous mutated genotype (G/G) carried by a baby. However, collagen X deficiency in mice (col10a1−/− mice) could be born, though the null mutations have phenotypic consequences which partially resemble MCDS in human (Kwan et al. 1997). The reason might be that the late onset of coxa vara and the relatively milder phenotypic changes in collagen X mutant mice (Kwan et al. 1997). One of the underlying mechanisms of MCDS could be haploinsufficiency that one mutated allele lead to an abnormal function copy of collagen X (Wallis et al. 1994; Warman et al. 1993). An alternative explanation of the SMD phenotype is that the truncated α1(X) chain collagen chains cause a dominant negative effect because of the deposition of abnormal collagen X in the matrix (Kwan et al. 1997).
Development of genomic technology greatly facilitate human disease mapping, and now it is feasible to locate disease-associated alleles by WGS, which allows examination of SNVs, indels, structure variations (SVs) and CNVs in coding and non-coding regions of the genome, without omitting regulatory regions, such as promoters and enhancers (Meienberg et al. 2016). In addition to SNPs, we examine other variant forms but failed to identify other promising variants associated with this disorder. One exception was that we identified a ~ 88 kb deletion (81134201–81222500) on chromosome 16 overlapping gene PKD1L2 in some affected family members, and it was absent from two healthy individuals. Moreover, there is no such CNV in the 1000 Genomes Project phase 3 data or in DGV database. PKD1L2 seems to be a polymorphic pseudogene in human and encodes one member of the polycystin protein family. The protein may function as a component of cation channel pores. But no study report that PKD1L2 is associated with MCDS, although one study (Mackenzie et al. 2009) reveals that upregulation of PKD1L2 cause neuromuscular disease in mouse and short form of PKD1L2 in human is expressed in the developing and adult heart as well as kidney (Yuasa et al. 2004). Nonetheless, the deletion only presents in four patients (two homozygous deletions and two heterozygous deletions), which thus could not explain the shared drawf status of all 17 affected individuals.
Taken together, the missense variant c.2011T>C (p.Ser671Pro) in COL10A1 is linked to the dwarfism in this Pakistan family. It is one of the 12 variants that are absent from global populations and highly conserved in evolution, indicating its functional importance despite its allelic heterogeneity. Our study also demonstrated that WGS is a powerful approach to determine functional variants associated with Mendelian disorders, which offers complete coverage of the coding region of the genome and allows examination of variants in non-coding regions.
Web resources
1000 Genomes GRCh37 human reference genome, ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/human_g1k_v37.fasta.gz
1000 Genomes Project phase 3 data, http://www.1000genomes.org/data
CADD, http://cadd.gs.washington.edu
ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/
Database of Genomic Variants (DGV), http://dgv.tcag.ca/dgv/app/home
ExAC Browser, http://exac.broadinstitute.org/
GERP, http://mendel.stanford.edu/SidowLab/downloads/gerp/
NCBI gene database, https://www.ncbi.nlm.nih.gov/gene/
NCBI structure database, https://www.ncbi.nlm.nih.gov/Structure/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Phylop, http://ccg.vital-it.ch/mga/hg19/phylop/phylop.html
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer 3.0, http://primer3.ut.ee
R version 3.2.1, http://www.r-project.org/
SIFT, http://sift.jcvi.org/
UCSC genome browser, http://genome.ucsc.edu/
Variant Effect Predictor (VEP), http://www.ensembl.org/info/docs/tools/vep/index.html
Data access
The accession number for the whole-genome sequences reported in this paper is National Omics Data Encyclopedia (NODE) (http://www.biosino.org/node): NODEP00000213.
References
Abyzov A, Urban AE, Snyder M, Gerstein M (2011) CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21(6):974–984
Bahcall OG (2016) Genetic variation: ExAC boosts clinical variant interpretation in rare diseases. Nat Rev Genet 17(10):584
Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA et al (2011) Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12(11):745–755
Bateman JF, Wilson R, Freddi S, Lamande SR, Savarirayan R (2005) Mutations of COL10A1 in Schmid metaphyseal chondrodysplasia. Hum Mutat 25(6):525–534
Chan Y, Salem RM, Hsu YH, McMahon G, Pers TH, Vedantam S et al (2015) Genome-wide analysis of body proportion classifies height-associated variants by mechanism of action and implicates genes important for skeletal development. Am J Hum Genet 96(5):695–708
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C et al (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498
1000 Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM et al (2015) A global reference for human genetic variation. Nature 526(7571):68–74
He M, Xu M, Zhang B, Liang J, Chen P, Lee JY et al (2015) Meta-analysis of genome-wide association studies of adult height in East Asians identifies 17 novel loci. Hum Mol Genet 24(6):1791–1800
Higuchi S, Takagi M, Shimomura S, Nishimura G, Hasegawa Y (2016) A Japanese familial case of Schmid metaphyseal chondrodysplasia with a novel mutation in COL10A1. Clin Pediatr Endocrinol 25(3):107–110
Ho MS, Tsang KY, Lo RL, Susic M, Makitie O, Chan TW et al (2007) COL10A1 nonsense and frame-shift mutations have a gain-of-function effect on the growth plate in human and mouse metaphyseal chondrodysplasia type Schmid. Hum Mol Genet 16(10):1201–1215
Ikegawa S, Nakamura K, Nagano A, Haga N, Nakamura Y (1997) Mutations in the N-terminal globular domain of the type X collagen gene (COL10A1) in patients with Schmid metaphyseal chondrodysplasia. Hum Mutat 9(2):131–135
Ikegawa S, Nishimura G, Nagai T, Hasegawa T, Ohashi H, Nakamura Y (1998) Mutation of the type X collagen gene (COL10A1) causes spondylometaphyseal dysplasia. Am J Hum Genet 63(6):1659–1662
Koch L (2014) Disease genetics: insights into missing heritability. Nat Rev Genet 15(4):218
Kwan KM, Pang MK, Zhou S, Cowan SK, Kong RY, Pfordte T et al (1997) Abnormal compartmentalization of cartilage matrix components in mice lacking collagen X: implications for function. J Cell Biol 136(2):459–471
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595
Lu D, Lou H, Yuan K, Wang X, Wang Y, Zhang C et al (2016) Ancestral origins and genetic history of Tibetan Highlanders. Am J Hum Genet 99(3):580–594
Mackenzie FE, Romero R, Williams D, Gillingwater T, Hilton H, Dick J et al (2009) Upregulation of PKD1L2 provokes a complex neuromuscular disease in the mouse. Hum Mol Genet 18(19):3553–3566
Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F et al (2016) The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538(7624):201–206
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ et al (2009) Finding the missing heritability of complex diseases. Nature 461(7265):747–753
McIntosh I, Abbott MH, Warman ML, Olsen BR, Francomano CA (1994) Additional mutations of type X collagen confirm COL10A1 as the Schmid metaphyseal chondrodysplasia locus. Hum Mol Genet 3(2):303–307
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A et al (2016) The Ensembl Variant Effect Predictor. Genome Biol 17(1):122
Meienberg J, Bruggmann R, Oexle K, Matyas G (2016) Clinical sequencing: is WGS the better WES? Hum Genet 135(3):359–362
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM et al (2010) Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42(1):30–35
Pritchard JK, Cox NJ (2002) The allelic architecture of human disease genes: common disease-common variant…or not? Hum Mol Genet 11(20):2417–2423
Schmid TM, Linsenmayer TF (1985) Immunohistochemical localization of short chain cartilage collagen (type X) in avian tissues. J Cell Biol 100(2):598–605
Stratakis CA, Orban Z, Burns AL, Vottero A, Mitsiades CS, Marx SJ et al (1996) Dideoxyfingerprinting (ddF) analysis of the type X collagen gene (COL10A1) and identification of a novel mutation (S671P) in a kindred with Schmid metaphyseal chondrodysplasia. Biochem Mol Med 59(2):112–117
Wallis GA, Rash B, Sweetman WA, Thomas JT, Super M, Evans G et al (1994) Amino acid substitutions of conserved residues in the carboxyl-terminal domain of the alpha 1(X) chain of type X collagen occur in two unrelated families with metaphyseal chondrodysplasia type Schmid. Am J Hum Genet 54(2):169–178
Wallis GA, Rash B, Sykes B, Bonaventure J, Maroteaux P, Zabel B et al (1996) Mutations within the gene encoding the alpha 1 (X) chain of type X collagen (COL10A1) cause metaphyseal chondrodysplasia type Schmid but not several other forms of metaphyseal chondrodysplasia. J Med Genet 33(6):450–457
Warman ML, Abbott M, Apte SS, Hefferon T, McIntosh I, Cohn DH et al (1993) A type X collagen mutation causes Schmid metaphyseal chondrodysplasia. Nat Genet 5(1):79–82
Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S et al (2014) Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 46(11):1173–1186
Yuasa T, Takakura A, Denker BM, Venugopal B, Zhou J (2004) Polycystin-1L2 is a novel G-protein-binding protein. Genomics 84(1):126–138
Acknowledgements
SX acknowledges financial support from the Strategic Priority Research Program (XDB13040100) and Key Research Program of Frontier Sciences (QYZDJ-SSW-SYS009) of the Chinese Academy of Sciences (CAS), the National Natural Science Foundation of China (NSFC) grant (91331204, 91731303, 31771388, and 31711530221), the National Science Fund for Distinguished Young Scholars (31525014), the Program of Shanghai Academic Research Leader (16XD1404700), and the National Key Research and Development Program (2016YFC0906403); YL acknowledges support from NSFC grant (31501011) and Science and Technology Commission of Shanghai Municipality (STCSM) (14YF1406800); HL acknowledges support from NSFC grant (31601046) and STCSM grant (16YF1413900); SX is a Max-Planck Independent Research Group Leader and member of CAS Youth Innovation Promotion Association. SX also gratefully acknowledges the support of the National Program for Top-notch Young Innovative Talents of the “Wanren Jihua” Project. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank LetPub (www.letpub.com) and Dr Asifullah Khan for their linguistic assistance during the preparation of this manuscript.
Author contributions
S.X. conceived and designed the study and supervised the project. F.I., M.A., F.B., and I.B., contributed to sample and phenotype collection. Y.L. managed laboratory work. S.X. contributed reagents and materials. C.Z. developed pipeline for processing NGS data and performed variant calling analysis. H.L., R.F., and Z. W developed pipeline for structural variation analysis. C.Z. and J.L. analyzed the data. S.X., and C.Z. wrote the main paper. C.Z. and J.L. prepared the Supplementary Information. All authors discussed the results and implications and commented on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
C Zhang and J Liu contributed equally to this work.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Zhang, C., Liu, J., Iqbal, F. et al. A missense point mutation in COL10A1 identified with whole-genome deep sequencing in a 7-generation Pakistan dwarf family. Heredity 120, 83–89 (2018). https://doi.org/10.1038/s41437-017-0021-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-017-0021-6
