
Abstract
The House Crow (Corvus splendens) is a useful study system for investigating the genetic basis of adaptations underpinning successful range expansion. The species originates from the Indian subcontinent, but has successfully spread through a variety of thermal environments across Asia, Africa and Europe. Here, population mitogenomics was used to investigate the colonisation history and to test for signals of molecular selection on the mitochondrial genome. We sequenced the mitogenomes of 89 House Crows spanning four native and five invasive populations. A Bayesian dated phylogeny, based on the 13 mitochondrial protein-coding genes, supports a mid-Pleistocene (~630,000 years ago) divergence between the most distant genetic lineages. Phylogeographic patterns suggest that northern South Asia is the likely centre of origin for the species. Codon-based analyses of selection and assessments of changes in amino acid properties provide evidence of positive selection on the ND2 and ND5 genes against a background of purifying selection across the mitogenome. Protein homology modelling suggests that four amino acid substitutions inferred to be under positive selection may modulate coupling efficiency and proton translocation mediated by OXPHOS complex I. The identified substitutions are found within native House Crow lineages and ecological niche modelling predicts suitable climatic areas for the establishment of crow populations within the invasive range. Mitogenomic patterns in the invasive range of the species are more strongly associated with introduction history than climate. We speculate that invasions of the House Crow have been facilitated by standing genetic variation that accumulated due to diversifying selection within the native range.
Similar content being viewed by others


Introduction
Studies of invasive species provide an opportunity to address fundamental questions about the ecology and evolution of colonisation. Successful invaders must quickly establish viable populations by undergoing rapid proliferation and local dispersal, which may involve the development of novel behavioural or genetic adaptations to new environments (Merilä et al. 1996; Novak 2007; Schwartz et al. 2007; Rollins et al. 2016). Genetic changes that allow species to adapt to new environments can arise through rapid acquisition of novel mutations or from standing genetic variation. Natural selection in invaders mainly operates on standing genetic variation, resulting in selective sweeps that favour adaptive alleles over neutral or deleterious alleles (Barrett and Schluter 2008; Prentis et al. 2008; Bock et al. 2015). Despite this, most research on avian invasion genetics has concentrated on comparing genetic diversity between native and introduced populations (Baker 1992; Hawley et al. 2006; Schrey et al. 2011) and few studies have investigated the role of adaptive evolution during the invasion process (Liebl et al. 2015; Rollins et al. 2016).
Species introduced to new environments often experience different bioenergetic demands compared to their native habitats, for example due to differences in nutrient availability or climate (Das 2006). Thus adaptations in genes mediating mitochondrial energetics could confer fitness benefits and enhance invasion success. In animals, the vast majority of the chemical energy required for cellular function is provided by the oxidative phosphorylation system (OXPHOS) that operates in the mitochondrial inner membrane (Bar-Yaacov et al. 2012). The 13 proteins encoded by the mitochondrial genome (mitogenome) are transmembrane subunits of the four major enzyme complexes responsible for OXPHOS (complexes I, III, IV, V) (Tsukihara et al. 1996). It has been demonstrated that, across diverse metazoan taxa, candidate substitutions under positive selection are disproportionately located in the genes encoding OXPHOS complex I (NADH-ubiquinone oxidoreductase) (Garvin et al. 2014). This enzyme serves as the primary electron input into the mitochondrial respiratory chain and generates approximately 40% of the proton-motive force used for ATP synthesis (Nicholls and Ferguson 2014). In addition, it is the main site of cellular reactive oxygen species (ROS) production (Murphy 2009) and is implicated in heat production (Lowell and Spiegelman 2000). This means that even single residue changes in this complex, through modulating ATP, ROS, or heat production, can result in broad consequences for organismal fitness and ecological adaptation (Wolff et al. 2014).
Consistently, there are reports that positive selection of OXPHOS complex I is linked to environmental adaptation of avifauna. For example, in the broadly distributed Tachycineta swallows, three mitochondrial genes (ND2, ND5 and CYTB belonging to OXPHOS complexes I and III) contain regions that exhibit signatures of diversifying selection in a background of purifying selection (Stager et al. 2014). Likewise, positive selection in the same enzymes has been linked to parapatric mitochondrial divergence of the eastern yellow robin (Eopsaltria australis) (Morales et al. 2015). As the distribution of mitochondrial genotypes in the swallows and robin species is strongly correlated with climate, these studies speculated that thermally driven selection may have influenced population structure, facilitated by positive selection of the mitogenome resulting in differences in mitochondrial coupling efficiencies and in turn ratios of ATP to heat production. These examples also demonstrate that mitochondrial protein-coding genes constitute useful genetic markers for investigating signatures of natural selection during episodes of adaptive evolution into novel environments. Indeed, mitochondrial functions are recognised as of sufficiently great evolutionary and ecological significance that the term ‘mitonuclear ecology’ has been coined for their study (Hill 2015).
The House Crow, Corvus splendens (Corvidae), is a suitable model to study mitogenome-based adaptations in the context of invasion because it has established populations outside its natural range in a wide range of environments. Estimates from mitochondrial control region sequences indicate that the House Crow and its sister species, the jungle crow (Corvus macrorhynchos), most likely diverged during a period of climate change in the Pliocene (Haring et al. 2012). Its native range stretches throughout the Indian subcontinent, spanning the wet, semiarid and arid zones of the tropical and subtropical regions, to more temperate alpine regions such as Nepal and the highlands of India (Fig. 1). Due to deliberate introductions to Africa (Zanzibar) and South East Asia (Malaysia) in the late nineteenth century, as well as more recent inadvertent introductions (Wells 2007; Ryall 2016), the House Crow has become widespread in many Asian and African countries and has established viable populations in a range of climatic zones (Fig. 1). A breeding population has recently become established even in considerably colder temperate climate areas in Western Europe, such as the Netherlands (Ryall 2016).

Distribution of the House Crow within native and invasive range (blue triangles and red circles, respectively) and distribution of the collected samples (black crosses) mapped over a Köppen–Geiger climate classification map (Kottek et al. 2006). Species distribution sourced from the Invasive Species Compendium (http://www.cabi.org/isc/datasheet/15463). Circles over sampling sites indicate which clade on a phylogenetic tree (Fig. 2) the samples belong to, dashed line indicates native north clade while solid line indicates native south clade
In this study, we investigated evolutionary history of the House Crow mitogenome, including history of lineage divergence and signals of natural selection. Because the native range of the House Crow is geographically large and encompasses diverse environments including climates, we expect genetic variation that is positively selected for local environments to pre-adapt invading lineages to thrive in similar environments in the invaded range. Under a scenario where positively selected variation in the native range facilitated invasion, we expect variants that are associated with a particular environment and show signatures of positive selection to be disproportionately represented in invaded populations with climates similar to that of the native source of those variants. To test this hypothesis, we sequenced the complete mitochondrial genomes from 89 House Crows representing four native and five invasive populations occupying diverse environments in Asia and Africa (Fig. 1). We used these data to investigate the diversity among House Crow lineages and estimate the time of mitochondrial DNA divergence among major lineages and determine the centre of radiation for the House Crow to better understand invasion pathways. We also tested for evidence of positive selection acting on OXPHOS genes and applied protein modelling to inform whether this could plausibly be linked to thermal selection of populations within the native range of the species. In addition, we used ecological niche modelling to predict the species distribution and investigate which of the climatic variables can be the best predictor of the House Crow distribution.
Materials and methods
Population sampling
A total of 89 House Crows were collected, representing six Asian (Malaysia, Sri Lanka, Bangladesh, India, Nepal, Singapore) and three African (Kenya, Tanzania, South Africa) populations within the species’ native and introduced ranges (Fig. 1). Sampling included sites in humid equatorial climates with a variable duration of dry season, warm temperate climates with warm and hot summers, and semiarid and arid climates (Fig. 1). Among sampled locations, the coolest was Nepal and South Africa, while the warmest locations included tropical Sri Lanka (native range) and Malaysia (invasive range). South Africa was characterised by the lowest average annual precipitation, while Malaysia had the highest average annual rainfall (Supplementary Material 1). All specimens were collected between February 2013 and December 2015. Samples included liver tissue obtained from bird carcasses, feathers plucked from trapped or dead birds, and dry shed feathers collected in the field (Supplementary Material 2). Muscle and liver tissue from two preserved House Crow specimens collected from the port of Karratha, Western Australia, but of unknown origin, were provided by Western Australia Museum. A sample originating from North India was contributed by the Australian National Wildlife Collection (toepad scrapings from birds collected in 1949). Western Australia Museum provided two House Crow specimens obtained from birds shot on a ship that arrived at Rottnest Island, Western Australia from India, which are thus assumed to be of Indian origin.
DNA extraction
DNA was extracted from tissue samples using the DNeasy Blood and Tissue kit (QIAGEN) with slight modifications introduced to the manufacturer’s protocol depending on the sample type (details in Supplementary Material 3). DNA extractions were conducted in a laboratory free of concentrated PCR products. Negative controls were included in each extraction to monitor cross-sample contamination. DNA was quantified using a Qubit fluorometer (ThermoFisher Scientific). The concentration of DNA obtained from non-invasive samples (feathers, museum specimens) ranged from 0.5 to 20 ng/µl, while from muscle and liver tissue from 15 to 200 ng/µl. DNA concentration for all the samples was optimised before further analyses were performed.
Mitogenome sequencing, assembly and annotation
Depending on the quality of extracted DNA, two methods of mitogenome sequencing were applied (Supplementary Material 2). Degraded DNA samples (n = 31) were subjected to whole-genome shotgun sequencing (2 × 150 bp paired-end run) run on the MiSeq Personal Sequencer (Illumina, San Diego, CA). Samples with high molecular weight DNA (n = 58) were subjected to long-range PCR amplification of mitochondrial genome prior to MiSeq sequencing. Long-range PCR (lrPCR) species-specific primers were designed to amplify complete mitogenomes in four overlapping fragments, each around 4.5 kb long and overlapping with the neighbouring fragments by at least 300 bp (Supplementary Material 4).
Mitogenome sequence assembly and annotation were performed following Gan et al. (2014) with slight modification for some sequences. Poorly assembling sequences, due to differential efficiency of PCR reactions causing highly variable coverage among PCR fragments, were subject to reconstruction of the mitochondrial genome using a two-step procedure that involved MIRA 3.4.1.1 and MITObim 1.6 assembly (Hahn et al. 2013). The mean sequencing coverage obtained for all the samples was ~65× with the minimum coverage of 6.5× (Supplementary Material 2). All complete mitochondrial genome sequences were deposited in GenBank (accessions KJ766304, KP019935-KP019940, KY018606-KY018687, KY050718). Consistency between the two sequencing approaches (lrPCR and shotgun sequencing) was ensured by comparing gene order and content of mitogenome assemblies of five randomly chosen samples sequenced using both methods (Supplementary Material 2). All assembled mitogenomes were blasted against the NCBI nucleotide database (with default parameters) and returned the highest match against the House Crow reference mitogenome KJ766304 (99% similarity and 100% query coverage).
Patterns of genetic diversity among lineages
Sequences of 13 mitochondrial DNA protein-coding genes were aligned and edited using Geneious 7.1.9 (Kearse et al. 2012). Out of 89 sequences, 44 unique haplotypes were identified. No indels were detected. Stop codons were removed, the ND6 gene sequence was reverse-complemented, and overlapping gene sequences were repeated to include the complete coding sequence for each gene. Genetic distances between haplotypes were calculated with Geneious 7.1.9. Standard genetic diversity metrics for each of the 13 protein-coding genes and complete mitochondrial genome were calculated. DnaSP v.5 (Librado and Rozas 2009) was used to calculate number of haplotypes (Nh), haplotype diversity (Hd), and nucleotide diversity (π). Arlequin 3.5.1.3 (Excoffier and Lischer 2010) was used to perform Tajima’s D (Tajima 1996) neutrality tests. A minimum spanning network among sequences (n = 89) was calculated with Arlequin and visualised with Cytoscape 3.2.1 (Shannon et al. 2003). Genetic diversity calculations were performed on a dataset where populations were defined based on geographical proximity: two populations in the native range, ‘native north’ (Nepal, n = 5; Bangladesh, n = 5; India, n = 3) and ‘native south’ (Sri Lanka, n = 8), and three populations in the invasive range, ‘East Africa’ (Tanzania, n = 27 and Kenya, n = 5), ‘South Africa’ (n = 27) and ‘South East Asia’ (Singapore, n = 6; Malaysia, n = 8).
Phylogenetic analyses
To infer phylogenetic relationships among House Crow haplotypes, we performed Bayesian phylogenetic analysis based on 13 protein-coding genes and complete mitochondrial genome sequences, together with a minimum spanning network analysis using the mitochondrial haplotypes. PartitionFinder (Lanfear et al. 2012) was run prior to phylogenetic analysis to find the most supported data partitioning and associated substitution models based on the Bayesian Information Criterion (BIC). Seven partitions were identified for the alignment of 13 protein-coding genes and for the complete mitochondrial genome (Supplementary Material 5). The intraspecific phylogenetic tree was inferred using the Bayesian approach implemented in MrBayes 3.2 (Ronquist et al. 2012) with the jungle crow (sister species) as an outgroup (NC027173.1) (Krzemińska et al. 2016b). Four independent runs were performed for 10 million generations and sampled every 1000 generations. Convergence of replicates was verified in TRACER v. 1.6 (Rambaut et al. 2014) by ensuring unimodality of posterior distributions and effective sample size (ESS > 200) for all parameters. Trees were summarised using the 50% majority rule method, after discarding the first 25% of the sample as burnin, and visualised using FIGTREE 1.4.2 (Rambaut 2012).
Estimating time to most recent common ancestor of the House Crow—interspecific analysis
Based on a broad scale Corvidae study that estimated divergence time for over 40 species, using two nuclear and two mitochondrial genes, it has been proposed that the genus Corvus has a Palaearctic origin and began to diversify in the mid-Miocene (~ 17.5 mya) (Jønsson et al. 2012). Here, interspecific analysis was conducted to estimate time to the most recent common ancestor (TMRCA) for the House Crow and the resulting estimate was then used in a separate analysis as a prior to assess divergence between House Crow lineages. First, a phylogenetic tree was built from an interspecific dataset comprising 13 mitochondrial protein-coding gene sequences for 18 avian species (Supplementary Material 6), including the two most divergent House Crow lineages (0.49% difference based on genetic distance, data not shown). Avian mitochondrial genomes included all sequences available for the family Corvidae, including the genus Corvus. Additional bird sequences, including Palaeognathae (common ostrich and white throated tinamou), Galloanseres (chicken and mallard) and Neoaves (zebra finch), were used from species where fossil records are available and can provide time constraints in our phylogenetic analysis of divergence. The minimum age constraint for the fossil records was adopted from Jarvis et al. (2014) while the maximum age constraint for all the fossil records was placed on the divergence between Neognathae and Palaeognathae which is the maximum age for the root node (99.6 my): TMRCA of Palaeognathae + Neognathae is 66–99.6 my (million years), of Struthio + Tinamus 20–99.6 my, of Anas + Gallus 51–99.6 my, of Corvidae + Taeniopygia 7.2–99.6 my (see Supplementary Material 7 for details of the origin and reference of each fossil record used in the analysis).
Interspecific BEAST analysis was run using first and second codon positions of mtDNA protein-coding genes, with third codon sites removed to reduce the impact of mutational saturation in the analysis. Nonetheless, divergence estimates based on the first and second codon positions overlapped with estimates based on the first, second and third codon positions for most nodes (Supplementary Material 8: Figs. 1 and 2). Linked substitution and clock models were used with a tree modelled with Birth Death process (Yule process was rejected based on the Bayes Factors, data not shown). As there was no evidence of rate heterogeneity among branches (a relaxed clock model was rejected by a preliminary run where the posterior distribution of coefficientOfVariation was close to 0, data not shown), we ran the analysis using a strict clock model with GTR+I+G nucleotide substitution model and fossil record time constraints included as priors with uniform distributions. The substitution model was assigned by comparing values of the BIC as implemented in PartitionFinder (Lanfear et al. 2012). The analysis was run for 100 million generations and sampled every 10,000 steps.
Estimating time of divergence of House Crow lineages—intraspecific analysis
Intraspecific BEAST analysis was performed (using estimates from the previous step) to estimate divergence between all tested House Crow lineages. In total, 44 unique House Crow mtDNA haplotypes were included in this analysis. The prior for the tree root was specified as TMRCA of two House Crow lineages estimated in the interspecific analysis (0.336–1.053 my). To ensure that first and second codon positions, which are more likely to be affected by negative selection, are not violating clock assumption (Subramanian et al. 2009), we ran two analyses: one with all three codon sites and one with only third codon sites included (Supplementary Material 8 and Figs. 3 and 4). We used the substitution model TIM + G as selected by PartitionFinder, a strict clock model (a relaxed clock model was rejected by a preliminary run where the posterior distribution of coefficientOfVariation was close to 0, data not shown) and a coalescent tree prior with constant size. The analysis was run for 100 million generations and sampled every 10,000 steps.
Detecting signatures of positive selection using a codon-based approach
Codon-based models of molecular evolution allow the inference of selection from alignments of homologous sequences by estimating the relative rates of synonymous (dS) and non-synonymous substitutions (dN) (Nielsen and Yang 1998). The ratio of non-synonymous to synonymous substitution (ω = dN/dS) is used as a measure of selective pressure, with ω > 1 indicating positive selection, ω < 1 indicating purifying selection and ω = 1 indicating neutrality (Nielsen and Yang 1998). Methods based on determining the average ω for a particular gene or region are likely to find an overwhelming signal of purifying selection, and can therefore mask positive selection acting on individual codons. For this reason, phylogeny-based models that use codons instead of complete genes/genomes are preferably used to detect positive selection (Meiklejohn et al. 2007, da Fonseca et al. 2008). In this study, the program HyPhy implemented in the web-server Datamonkey (Delport et al. 2010) was used to identify codons putatively under selection using Fast Unconstrained Bayesian AppRoximation (FUBAR) (Murrell et al. 2013) and Mixed Effects Model of Evolution (MEME) (Murrell et al. 2012). FUBAR assigns each codon a posterior probability (PP) of belonging to three classes of ω: ω < 1, ω = 1 and ω > 1. Codons with PP > 0.9 and ω > 1 or ω < 1 were inferred to have evolved under positive or purifying selection, respectively. While FUBAR identifies codons putatively under selection across all branches on the phylogeny, MEME allows ω at each codon to vary across branches/lineages and so detects episodic selection (i.e. positive selection that varies temporally across branches/lineages). We assumed codons with p < 0.05 to be affected by the episodic positive selection. A phylogenetic tree built with MrBayes using 13 protein-coding genes was used (without the outgroup) for the codon-based selection analyses and subsequent analysis based on amino acid properties.
Detecting selection of amino acid properties based on a phylogenetic tree
A codon-based approach is sensitive to sampling strategy, because closely related individuals will have low sequence divergence and therefore the ω ratio will be relatively low. To supplement the codon-based approach, we used TreeSAAP (Woolley et al. 2003) to estimate significant changes in the amino acid properties along the phylogeny. TreeSAAP assesses the physicochemical effect of amino acid replacements across the phylogeny. The distributions of 20 physicochemical amino acid properties are analysed under the assumption that under neutrality, every feasible amino acid replacement is equally likely. Physicochemical properties with reported low accuracies (<70%) were not considered in order to avoid false positives (McClellan and Ellison 2010). We ran the analysis with a sliding window size of 10 codons, in 1-codon increments. Only amino acid replacements with magnitude categories ≥6 and z-scores above 3.09 and below −3.09 (p < 0.001) were considered as potentially under positive or purifying selection, respectively, as recommended by McClellan and Ellison (2010). Only differences that mapped to the internal branches on the tree were classified as true candidate variants under positive selection, because differences at the terminal tips could represent cases in which purifying selection has not yet purged deleterious forms (Rand et al. 2004). To ensure that the inclusion of genes potentially under positive selection does not bias our estimates of the tree, the intraspecies analysis in BEAST was re-run excluding genes detected to be under positive selection (described in the following section); no significant changes in tree estimates were observed (Supplementary Material 8: Figs. 3 and 4).
Protein structural analysis
The predicted structures of crow OXPHOS complex I protein variants identified as under positive selection were estimated using intensive homology modelling. Homology models were created in the I-TASSER server (Zhang 2008) using the 4.3 Å structure of bovine complex I solved by cryo-electron microscopy (cryo-EM) (Zhu et al. 2016) as the principal template. The four models obtained were of good quality with C-scores > 0.60 and TM-scores > 0.80. Protein structure was visualised in UCSF Chimera (Pettersen et al. 2004). These models are useful to identify amino acids that are plausible candidates for involvement in thermal adaptation (Sunnucks et al. 2017). Homology models give insights into whether particular amino acid substitutions are likely to impact mitochondrial ‘coupling’ efficiency (the ratio of adenosine triphosphate synthesis per unit substrate and O2 consumed), which is hypothesised to be closely linked to adaptive thermal biology.
Testing the invasive potential of native-range House Crows via ecological niche models
House Crow occurrence points in the form of unique latitude–longitude combinations were gathered through searching eBird observation records. Ecological variables summarising aspects of temperature and precipitation variation over the latter half of the twentieth century were used in the form of bioclimatic GIS layers at a spatial resolution of 10 km (Hijmans et al. 2005). It has been documented that several of the bioclimatic variables can be correlated (Peterson 2007; Peterson and Nakazawa 2008), and therefore we initially explored using a subset of seven environmental variables previously identified as having good predictive potential within the region that includes House Crow native and invasive ranges (Reddy and Nyári 2015). Additionally, a test for correlation of these bioclimatic variables was carried out in SDMToolbox in ArcMap (Brown 2014), and identified as suitable (<0.80 absolute correlation, data not shown) only four variables: annual mean temperature (Bio1), mean diurnal range (Bio2), annual precipitation (Bio12) and precipitation of the driest month (Bio14). Ecological niche models were constructed via MaxEnt v.3.4.1 (Phillips et al. 2006), by first developing models for the species’ native range and then projecting these conditions onto the global scale to infer potential climatic suitability throughout the invaded range (Nyári et al. 2006). The spatial extent of the native range, as well as the area and climatic space accessible for the species (Barve et al. 2011), was set based on a rectangle encompassing the four extreme most edges of all House Crow occurrence points. We ran MaxEnt using standard settings, setting aside 25% of occurrence points for model testing.
Results
Genetic diversity and divergence between House Crow lineages
A total of 89 House Crows were collected from nine different populations spanning a range of climates and land types (Fig. 1). The mitogenomes of all the birds were successfully sequenced and the consistency between shotgun sequencing and long-range PCR sequencing was confirmed with 100% sequence identity for the five selected individuals (Supplementary Material 2). There was evidence of heteroplasmy for three individuals (due to high mean sequencing coverage (~65×), differing variants were more likely to be signatures of heteroplasmy rather than sequencing errors), with two variants located in non-coding regions of the mtDNA (control region) and one within tRNA-Glu encoding sequence; this heteroplasmy did not influence the subsequent analyses, which focused on the 13 protein-coding genes. For the analyses where complete mitogenome sequences were used (diversity measures), the most common variant was used.
Based on alignments of the 13 protein-coding mitochondrial genes, 44 unique haplotypes were identified (Supplementary Material 9). The overall nucleotide diversity across mitochondrial genes was low (0.0016), with the lowest diversity observed for the ATP8 gene (0.00054) and the highest diversity for ND2 (0.00389) (Supplementary Material 10). Haplotype diversity and nucleotide diversity were highest for populations within the native range (India, Nepal, Bangladesh and Sri Lanka; Table 1). Among invasive populations, the highest haplotype and nucleotide diversity were observed for Singapore (Supplementary Material 11). Minimum spanning network analysis revealed patterns of haplotype sharing among populations in Malaysia, Singapore and Sri Lanka and no shared haplotypes between populations in Africa (Kenya, Tanzania, South Africa) (Supplementary Material 11).
Interspecific and intraspecific phylogenetic analyses using MrBayes and BEAST
We used interspecific and intraspecific analyses to gain evolutionary insights into the origin and diversification history of the House Crow lineages. Our divergence analysis supports the conclusion that the House Crow separated from its sister species jungle crow in the mid-Pliocene (Zanclean stage) at ~4.1 mya (3.06–5.20 my). Phylogenetic relationships between representatives of the family Corvidae and the genus Corvus were consistent with previous phylogenetic studies (Haring et al. 2012; Jønsson et al. 2012; Krzemińska et al. 2016b). Interspecific and intraspecific divergence estimates and tree topologies using all three codon positions and either first and second or only third codon positions were comparable (Supplementary Material 8).
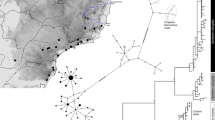
MrBayes and BEAST analyses resulted in similar tree topologies (Fig. 2a, b). The basal position of lineages from the northern part of the native range relative to the jungle crow outgroup supports a northern South Asian (Bangladesh, Nepal and possibly Northern India) origin for the species (Fig. 2a, b) dated at ~0.63 mya (0.34–0.99 my). Clade 1 (Fig. 2a) includes lineages from native north and native south range. Further evolution of maternal lineages resulted in one of the ancestral lineages (Clade 2) splitting into two at ~0.44 mya (0.20–0.76). One of the lineages remained in the mainland (Clade 3), while another dispersed to Sri Lanka (Clade 4), which has been physically separated from India for more than 10 my (Pethiyagoda 2012). This ~0.44 mya divergence event is also the estimate of TMRCA of all analysed invasive range lineages.

a Rooted phylogenetic summary tree (50% majority rule) estimated using Bayesian inference in MrBayes. All non-collapsed nodes were well supported (PP > 0.9). Main Clades are numbered and indicated with black circles. The candidate variants under positive selection: V159I in ND2 and T117A, A526T and V489I in ND5 gene indicated for relevant branches. The outgroup (not shown) was Corvus macrorhynchos. b BEAST dated phylogenetic tree where numbers show mean (and 95% highest posterior densities) estimates of divergene time between lineages (in million years). Both trees inferred based on 13 protein-coding genes representing House Crow lineages originating from different locations; terminal tips colour coded based on the geographical origin of individuals. Native range indicated by squares with diagonal lines. Very short branches within species clades have been collapsed for better presentation; triangle height corresponds to number of sequences and triangle width corresponds to nucleotide diversity within clade
Most invasive populations (Kenya, Singapore, Malaysia, South Africa, Tanzania) and the samples of unknown origin cluster with Sri Lanka lineages (Clade 4). A subset of samples in Tanzania and Singapore clustered with native north Indian lineages (Clade 3). Temperate climate South Africa and tropical climate South East Asian populations clustered with Sri Lanka lineages (native south). No invasive population shared haplotypes with the native lineages from colder climates (Nepal).
Natural selection analyses
We next used codon-based approaches to detect signals of diversifying and purifying selection acting on the House Crow mitogenome. All models revealed evidence of strong, pervasive purifying selection acting across all mitochondrial protein-coding genes (with posterior probability, PP > 0.9; results not shown). FUBAR analysis provided evidence of diversifying selection (PP > 0.9) at three codons in ND5 (Table 2) and MEME analysis suggested episodic diversifying selection acting at one codon (p < 0.05) in COI (Table 2). TreeSAAP analysis (Table 2 and Supplementary Material 10) provided evidence for positive selection of significant changes in alpha-helical tendencies and ionisation constants across seven OXPHOS subunits, including in amino acids implicated by the codon-based analyses (Table 2 and Supplementary Material 12). However, only four amino acid replacements in two genes were significant (p < 0.001) and located at the internal branches, namely V159I of ND2 and T117A, A526T and V489I of ND5. The ND2 V159I substitution was fixed in Clade 1, comprising all native and invasive lineages except for several lineages in Bangladesh and Nepal (top branch; Fig. 2a). Given the broad distribution of this mutation it is unlikely to be selected for climatic conditions. The ND5 T117A replacement was fixed in Clade 2, which comprised the native north, native south and invasive lineages. Again, this mutation appears to be too widely distributed to be selected for climatic conditions. The amino acid change V489I within ND5 was fixed within Clade 5 that includes the native Sri Lanka and four of five invasive populations (Malaysia, Singapore, Kenya, Tanzania). The amino acid change A526T within ND5 occurred in the branch leading to Clade 3 that includes native north lineages (Nepal, Bangladesh, India) and Singapore and Tanzania from the invasive ranges (Table 2 and Fig. 2a). These last two mutations, A526T and V489I, may represent independent adaptations to environments in the native north and south ranges, respectively (Fig. 1).
Structural and mechanistic effects of OXPHOS complex I variants
We next investigated the effects of the four substitutions inferred to be under positive selection on the structure and function of OXPHOS complex I. For this, homology models of the crow ND2 and ND5 variants were constructed and mapped to the complete cryo-EM structure of ovine complex I (Fiedorczuk et al. 2016) (Supplementary Material 13). Superimposition of the ND5 variants suggests that the non-conservative ND5 substitutions T117A and A526T may influence the tertiary structure of the subunit and in turn could modulate the coupling efficiency of complex I (Figs. 3a). This in turn supports the inferences of the TreeSAAP analysis (Table 2). Substitution at residue 526 of ND5 occurs at the start of helix HL, a hinge-like region predicted to be critical for propagating conformational changes between the proton channels encoded by ND2, ND4 and ND5 (Sato et al. 2013). Homology models constructed with I-TASSER suggests that this mutation influences the coiling of helix HL in crow ND5 variants (Fig. 3b), and similar effects were predicted with homology modelling using Phyre2, RaptorX and SWISS-MODEL (data not shown). This region is the most commonly observed site of positive selection in the mitogenomes of diverse animal species (Garvin et al. 2016).

Structural and mechanistic effects of OXPHOS complex I polymorphisms observed between House Crow lineages. a The structures of the seven mitochondrially encoded OXPHOS complex I subunits are shown based on the complete 3.9 Å structure of ovine complex I (Fiedorczuk et al. 2016). The cryo-EM structures of ovine ND1, ND3, ND4, ND4L and ND6 are shown in grey. The homology-modelled structures of crow ND2 and ND5 protein variants are shown in blue (ND2 V159 and ND5 T117, V489, A526) and yellow (ND2 I159 and ND5 A117, I489, T526). b Comparison of structures of ND5 T117, V489, A526 (top, blue) and ND5 A117, I489, T526 (bottom, yellow). c Comparison of structures of ND2 V159 (top, blue) and ND2 I159 (bottom, yellow). In b and c, the secondary structure, surface space/properties and substituted residues are shown
The homology models also suggest the other two substitutions could influence House Crow complex I function despite their conservative nature. The ND5 V489I substitution maps to helix TM15, which serves as a critical mediator of interactions between complex I and complex IV during mitochondrial respirasome formation (Wu et al. 2016) (Fig. 3b). In contrast, the ND2 V159I mutation occurs at the base of the ND2 proton efflux channel (Fiedorczuk et al. 2016; Zhu et al. 2016); surface modelling suggests that the additional methyl group occludes the channel and hence may partially restrict proton flow (Fig. 3c). However, it is presently not possible to make more detailed mechanistic predictions about if and how these substitutions will affect coupling due to the paucity of knowledge about residue-specific structure–function relationships in OXPHOS complex I.
Ecological niche models
We used ecological niche model to predict House Crow distribution and to identify climatic variables that are best predictors of the species’ distribution. In general, presence points exhibited a high density within urban areas, and had to be spatially subsampled to reduce spatial autocorrelation. A total of 3223 spatially unique House Crow occurrence points from the native range were at the basis of our modelling work. Of the bioclimatic variables, annual precipitation (Bio12) had the highest contribution towards the model, followed by annual mean temperature (Bio1), mean diurnal range (Bio2) and precipitation of driest month (Bio14). With few exceptions, the model predicted all known geographic areas where House Crows have, or have had, successful colonisation as having suitable bioclimatic niches for the species (Fig. 4)

Map representing suitable areas of House Crow invasion based on the MaxEnt model generated from bioclimatic variables of the species’ native range (red square) and projected onto a global scale. Hues of green represent lower suitability, while dark blue indicates highly suitable conditions. The model represents a thresholded output, allowing for a 10% omission error of occurrence points used in model training
.
Discussion
In this study, we used population mitogenomics to understand the evolutionary history of the House Crow and evolutionary drivers of divergence of its mitochondrial lineages. Previous studies have reported a Palearctic origin of genus Corvus (Jønsson et al. 2012), but there was no specific location indicated and there have been no intraspecific studies conducted to date. Our study is the first intraspecies dated phylogenetic analysis that provides insights into the House Crow’s evolutionary history. Our phylogenetic analyses suggest that northern South Asia (Nepal, Bangladesh and possibly North India) is the centre of origin for the House Crow (Fig. 2a). However, only a limited number of samples from the native range (particularly India) has been analysed here, thus our results should be interpreted with caution. We found that divergence among native range lineages started in mid-Pleistocene (~0.34–0.99 mya; Fig. 2b). Phylogenetic patterns and results of divergence analysis suggest that there is substantial geographic structure particularly between native north and native south clades. We found that the species thereafter diversified throughout South Asia and became invasive due to multiple introductions through Africa and South East Asia.
Most of the invasive range lineages sampled in this study clustered together with native south lineages (Clade 4). We therefore suggest lineages from Sri Lanka, or other closely related lineages not sampled in this study, as source populations for invasive range populations in Africa (South Africa, Kenya, Tanzania) and South East Asia (Malaysia, Singapore), as well as the two individuals at the port in Western Australia. Interestingly, a subset of Tanzanian haplotypes is also closely related to the Indian samples (Clade 3). This finding is consistent with the known history of the House Crow in Tanzania, where birds were brought to Zanzibar from the native range in India (Berruti 1997). However, there is a close relationship of some other Tanzanian haplotypes with the native south lineages (Fig. 2a and Supplementary Material 11) consistent with additional introductions from other locations (Sri Lanka or closely related) to Africa. The hypothesis of multiple introductions is further supported by the finding that samples collected from Mombasa, Kenya were of different haplotypes than those of neighbouring Tanzania (Clade 3 and Clade 5). Among invasive populations, Singapore was the most diverse, with several haplotypes clustering with different invasive populations (Fig. 2a and Supplementary Material 11). This may be directly linked to Singapore having one of busiest ports in the tropics, resulting in multiple opportunities for crows hitchhiking on ships to enter this country from different locations, leading in turn to a high level of population admixing (Krzemińska et al. 2016a). In contrast, there was surprisingly low haplotype diversity in South Africa (Table 1 and Supplementary Material 11), despite high levels of marine traffic in the region during the closure of the Suez Canal between 1967 and 1980 (Berruti 1997). This result may indicate either fewer introductions than expected or genetic bottlenecks caused by local pressures, for example the successful control programme recently launched in the region (Mfundo Tafeni, personal communication). Because mtDNA is a single gene prone to drift in small populations, sex-biased asymmetries and selection (Toews and Brelsford 2012), the mtDNA phylogeny presented here may not represent a history of population splitting; phylogenetic analyses based on multilocus nuclear sequencing data will be required to better understand history of population divergence.
We hypothesised that standing genetic variation present in the native range might have facilitated House Crow invasion. Consistently, we uncovered signatures of natural selection in the mitogenome within House Crow native and introduced ranges. The putative signatures of positive selection were associated with diversification of lineages within the native range, and we found no evidence that the species accumulated selectively beneficial mutations following introductions within invasive ranges. We therefore reject the hypothesis that native range lineages that are associated with a particular environment and show signatures of positive selection that may have facilitated invasion of habitats with similar climates. Because the signatures of positive selection are found in lineages within the native range and the ENM predicts currently invaded areas as suitable, we speculate that standing genetic variation within the native range might have pre-adapted the species to live in variable environments and consequently facilitated invasions. Overall, mitogenomic patterns in the invasive range of the species seem to be more strongly associated with introduction history than climate. With the exception of the Arabian Peninsula, ecological niche modelling predicted the suitability of all known geographic areas where House Crows have, or have had successful colonisations (Nyári et al. 2006; Ryall 2016), including invasive range areas tested in this study (Figs. 1 and 4). Ecological niche modelling suggests that precipitation may be important factor in predicting House Crow distribution.
Our genetic and biochemical analyses support selective diversification within the native range. Four substitutions in OXPHOS complex I showed evidence of positive molecular selection in the native range (Table 2 and Fig. 2a) and our atomic models suggest that each of these substitutions could plausibly affect coupling efficiencies of OXPHOS complex I. For example, ND2 V159I is predicted to directly modulate proton flow through the complex, and ND5 A526T may cause structural changes that affect energy propagation between subunits. We predict that such differences, through subtly modulating the ratios of proton translocation, heat generation and ROS production through complex I, may result in physiological differences that might be adaptive in different environments. For example, if the ND5 V489I and A526T substitutions modulate mitochondrial heat production, differential thermal selection pressures may favour their increased frequency in the tropical southern and alpine northern ranges, respectively. Likewise, changes in coupling efficiencies modulated by the ND2 V159I and ND5 T117A substitutions may have facilitated the evolution of basal clades into more environmentally adaptable ones. These findings contribute to the growing evidence that selection for OXPHOS traits has promoted genetic diversification of bird species (Morales et al. 2015; Stager et al. 2014). Recent studies indicate that mitolineage divergence could be accompanied by divergence of co-evolved nuclear genes with mitochondrial function (Garvin et al. 2016; Morales et al. 2016, 2017; Sunnucks et al. 2017). To determine whether this is the case in House Crow needs further study using genome-wide data.
Finally, we explored the observed relationship that most invasive populations are derived from native range Sri Lankan lineages and/or their unsampled relatives. One out of four detected signatures of positive selection, namely ND5 V489I, was located on the branches leading to native south Clade 5 (Fig. 2a). It is possible that the environmental conditions of the island have driven metabolic changes in these native lineages. Consistent with this suggestion, Sri Lanka has very diverse biogeography and climate (Fernando 1984). The wet zone of the island in the southwestern part (including the sampling site Colombo) is isolated from the nearest wet forests in the Indian mainland by dryer regions (Fernando 1984). Even during the Pleistocene period of land–bridge connections between India and Sri Lanka, the wet zones of these countries remained separate. As a result, species in southwestern Sri Lanka evolved in isolation, giving rise to the high levels of endemism evident today (Pethiyagoda 2012). Alternatively, given Sri Lanka is an island population, the ND5 V489I substitution could have arisen as a result of relaxed purifying selection, due to strong drift in small bottlenecked population, which would be expected during the invasion of the island.
Future research should involve testing crows from Arabian Peninsula. This region was not predicted as suitable by the ecological niche model, suggesting that House Crows might have evolved adaptations to much drier environment. In addition, future research should test whether variants under selection in the native range are adaptive in the invasive range. This could be achieved through physiological studies comparing cellular and organismal energetics of lineages for which selection has been detected (Sunnucks et al. 2017) and by including more samples from the native range, particularly India.
Data archiving
Sequence data have been deposited at GenBank under the accession numbers: KJ766304, KP019935-KP019940, KY018606-KY018687, KY050718.
References
Baker AJ (1992) Genetic and morphometric divergence in ancestral European and descendent New Zealand populations of chaffinches (Fringilla coelebs). Evolution 46:1784–1800
Bar-Yaacov D, Blumberg A, Mishmar D (2012) Mitochondrial-nuclear co-evolution and its effects on OXPHOS activity and regulation. Biochim Biophys Acta 1819:1107–1111
Barrett R, Schluter D (2008) Adaptation from standing genetic variation. Trends Ecol Evol 23:38–44
Barve N, Barve V, Jiménez-Valverde A, Lira-Noriega A, Maher SP, Peterson AT, Soberón J, Villalobos F (2011) The crucial role of the accessible area in ecological niche modeling and species distribution modeling. Ecol Model 222:1810–1819
Berruti A (1997) House crow. In: Harrison JA, Allan DG, Underhill LG, Herremans M, Tree AJ, Parker V, Brown CJ (eds) The Atlas of Southern African Birds. BirdLife South Africa, Johannesburg, South Africa, p 108
Bock DG, Caseys C, Cousens RD, Hahn MA, Heredia SM, Hübner S, Turner KG et al. (2015) What we still don’t know about invasion genetics. Mol Ecol 24:2277–2297
Brown J (2014) SDMtoolbox: a python-based GIS toolkit for landscape genetic, biogeographic and species distribution model analyses. Methods Ecol Evol 5:694–700
da Fonseca RR, Johnson WE, O’Brien SJ, Ramos MJ, Antunes A (2008) The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics 9:119
Das J (2006) The role of mitochondrial respiration in physiological and evolutionary adaptation. Bioessays 28:890–901
Delport W, Poon AF, Frost SD, Pond SLK (2010) Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567
Fernando CH (1984) Ecology and biogeography in Sri Lanka, 1st edn. Martinus Nijhoff Publishers, The Hague
Fiedorczuk K, Letts JA, Degliesposti G, Kaszuba K, Skehel M, Sazanov LA (2016) Atomic structure of the entire mammalian mitochondrial complex I. Nature 538:406–410
Gan HM, Schultz MB, Austin CM (2014) Integrated shotgun sequencing and bioinformatics pipeline allows ultra-fast mitogenome recovery and confirms substantial gene rearrangements in Australian freshwater crayfishes. BMC Evol Biol 14:19
Garvin MR, Bielawski JP, Sazanov LA, Gharrett AJ (2014) Review and meta-analysis of natural selection in mitochondrial complex I in metazoans. J Zool Syst Evol Res 53:1–17
Garvin MR, Templin WD, Gharrett AJ, DeCovich N, Kondzela CM, Guyon JR et al. (2016) Potentially adaptive mitochondrial haplotypes as a tool to identify divergent nuclear loci. Methods Ecol Evol. https://doi.org/10.1111/2041-210X.12698.
Hahn C, Bachmann L, Chevreux B (2013) Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res 41:e129
Haring E, Daubl B, Pinsker W, Kryukov A, Gamauf A (2012) Genetic divergences and intraspecific variation in corvids of the genus Corvus (Aves: Passeriformes: Corvidae)—a first survey based on museum specimens. J Zool Syst Evol Res 50:230–246
Hawley DM, Hanley D, Dhondt AA, Lovette IJ (2006) Molecular evidence for a founder effect in invasive house finch (Carpodacus mexicanus) populations experiencing an emergent disease epidemic. Mol Ecol 15:263–275
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978
Hill GE (2015) Mitonuclear ecology. Mol Biol Evol 32:1917–1927
Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C et al. (2014) Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 346:1320–1331
Jønsson KA, Fabre PH, Irestedt M (2012) Brains, tools, innovation and biogeography in crows and ravens. BMC Evol Biol 12:72
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kottek MJ, Grieser J, Beck C, Rudolf B, Rubel F (2006) World map of the Köppen-Geiger climate classification updated. Meteorol Z 15:259–263
Krzemińska U, Wilson R, Song BK, Seneviratne S, Akhteruzzaman S, Gruszczyńska J et al. (2016a) Genetic diversity of native and introduced populations of the invasive house crow (Corvus splendens) in Asia and Africa. Biol Inv 18:1867–1881
Krzemińska U, Wilson R, Rahman S, Song BK, Ming GH, Hua TM et al. (2016b) The complete mitochondrial genome of the invasive house crow Corvus splendens (Passeriformes: Corvidae). Mitochondrial DNA 27:974–975
Lanfear R, Calcott B, Ho SYW, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analysis. Mol Biol Evol 29:1695–1701
Librado P, Rozas J (2009) DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Liebl AL, Schrey AW, Andrew SC, Sheldon EL, Griffith SC (2015) Invasion genetics: lessons from a ubiquitous bird, the house sparrow Passer domesticus. Curr Zool 61:465–476
Lowell BB, Spiegelman BM (2000) Towards a molecular understanding of adaptive thermogenesis. Nature 404:652–660
McClellan DA, Ellison DD (2010) Assessing and improving the accuracy of detecting protein adaptation with the TreeSAAP analytical software. Int J Bioinform Res Appl 6:120–133
Meiklejohn CD, Montooth KL, Rand DM (2007) Positive and negative selection on the mitochondrial genome. Trends Genet 23:259–263
Merilä J, Björklund M, Baker AJ (1996) The successful founder: genetics of introduced Carduelis chloris (greenfinch) populations in New Zealand. Heredity 77:410–422
Morales HE, Pavlova A, Joseph L, Sunnucks P (2015) Positive and purifying selection in mitochondrial genomes of a bird with mitonuclear discordance. Mol Ecol 24:2820–2837
Morales HE, Pavlova A, Amos N, Major R, Bragg J, Kilian A et al. (2016) Mitochondrial-nuclear interactions maintain a deep mitochondrial split in the face of nuclear gene flow. bioRxiv. https://doi.org/10.1101/095596
Morales HE, Sunnucks P, Joseph L, Pavlova A (2017) Perpendicular axes of differentiation generated by mitochondrial introgresssion. Mol Ecol 26:3241–3255
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Murrell B, Moola S, Mabona A, Mabona A, Weighill T, Sheward D et al. (2013) FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol Biol Evol 30:1196–1205
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Pond SLK (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764
Nicholls DG, Ferguson SJ (2014) Bioenergetics, 4th edn. Academic Press:London
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Novak SJ (2007) The role of evolution in the invasion process. Proc Natl Acad Sci USA 104:3671–3672
Nyári ÁS, Ryall C, Peterson AT (2006) Global invasive potential of the house crow Corvus splendens based on ecological niche modelling. J Avian Biol 37:306–311
Peterson AT (2007) Why not WhyWhere: the need for more complex models of simpler environmental spaces. Ecol Model 203:527–530
Peterson AT, Nakazawa Y (2008) Environmental data sets matter in ecological niche modelling: an example with Solenopsis invicta and Solenopsis richteri. Global Ecol Biogeogr 17:135–144
Pethiyagoda R (2012) Biodiversity conservation in Sri Lanka’s novel ecosystems. Ceylon J Sci 41:1–10
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC et al. (2004) UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Model 190:231–259
Prentis PJ, Wilson JRU, Dormontt EE, Richardson DM, Lowe AJ (2008) Adaptive evolution in invasive species. Trends Plant Sci 13:288–294
Rambaut A, Suchard MA, Xie D, Drummond AJ (2014) Tracer v1.6. Available at: http://beast.community/tracer. Accessed 28 Oct 2016.
Rambaut A (2012) Figtreev1.4. 2. Available at: http://tree.bio.ed.ac.uk/software/figtree/. Accessed 20 November 2016.
Rand DM, Haney RA, Fry AJ (2004) Cytonuclear coevolution: the genomics of cooperation. Trends Ecol Evol 19:645–653
Reddy S, Nyári ÁS (2015) Novel insights into the historical biogeography of the streak-breasted scimitar babbler complex (Aves: Timaliidae: Pomatorhinus ruficollis complex). Curr Zool 61:910–921
Rollins LA, Woolnough AP, Fanson BG, Cummins ML, Crowley TM, Wilton AN et al. (2016) Selection on mitochondrial variants occurs between and within individuals in an expanding invasion. Mol Biol Evol 33:995–1007
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S et al. (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Ryall C (2016) House crow monitor Corvus splendens. Available at: http://www.housecrow.com. Accessed 11 Jul 2016.
Sato M, Sinha PK, Torres-Bacete J, Matsuno-Yagi A, Yagi T (2013) Energy transducing roles of antiporter-like subunits in E. coli NDH-1 with main focus on subunit NuoN (ND2). J Biol Chem 288:24705–24716
Schrey AW, Grispo M, Awad M, Cook MB, McCoy ED, Mushinsky HR et al. (2011) Broad-scale latitudinal patterns of genetic diversity among native European and introduced house sparrow (Passer domesticus) populations. Mol Ecol 20:1133–1143
Schwartz MK, Luikart G, Waples RS (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22:25–33
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Stager M, Cerasale DJ, Dor R, Winkler DW, Cheviron ZA (2014) Signatures of natural selection in the mitochondrial genomes of Tachycineta swallows and their implications for latitudinal patterns of the ‘pace of life’. Gene 546:104–111
Subramanian S, Denver DR, Millar CD, Heupink T, Aschrafi A, Emslie SD et al. (2009) High mitogenomic evolutionary rates and time dependency. Trends Cell Biol 25:482–486
Sunnucks P, Morales HE, Lamb AM, Pavlova A, Greening C (2017) Integrative approaches for studying mitochondrial and nuclear genome co-evolution in oxidative phosphorylation. Front Genet 8:25
Tajima F (1996) The amount of DNA polymorphism maintained in finite population when the neutral mutation rate varies among sites. Genetics 143:1457–1465.
Toews DPL, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21:3907–3930
Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K et al. (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science 272:1136–1144
Wells DR (2007) The birds of the Thai-Malay Peninsula: passerines, vol 2. Christopher Helm, London.
Wolff JN, Ladoukakis ED, Enríquez JA, Dowling DK (2014) Mitonuclear interactions: evolutionary consequences over multiple biological scales. Philos Trans R Soc B 369:20130443
Woolley S, Johnson J, Smith MJ, Crandall KA, McClellan DA (2003) TreeSAAP: selection on amino acid properties using phylogenetic trees. Bioinformatics 19:671–672
Wu M, Gu J, Guo R, Huang Y, Yang M (2016) Structure of mammalian respiratory supercomplex I1III2IV1. Cell 167:1598–1609
Zhu J, Vinothkumar KR, Hirst J (2016) Structure of mammalian respiratory complex I. Nature 536:354–358
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 23:9
Acknowledgements
We would like to acknowledge Agri-Food and Veterinary Authority in Singapore, Subang Jaya Council, Dr. Sampath Seneviratne from the University of Colombo, University of Dhaka, Mfundo Tafeni from the Environmental Resource Management Department in Cape Town, Duncan Mitchell from Kenya, the Assistant Veterinary Officer, Meor Amri Md. Noor from the Penang Council, David Alila from Tanzania, Australian National Wildlife Collection and Western Australia Museum for providing specimens for DNA testing. We also thank Monash University Malaysia Genomics Facility for generating data used in this project; Dr. Han Ming Gan and Tan Mun Hua for their assistance with bioinformatics. We are also extremely grateful to Dr Collin Ryall, Prof Simon Ho, and The Persistence and Adaptation Research Team (PART) for their help in parts of this study. Funding for this study was provided by the Monash University Malaysia School of Science and Monash University Malaysia Tropical Medicine Biology Multidisciplinary Platform and the internal Monash sources, including PART research group funds. We would also like to acknowledge the Australian Government for granting the Endeavour Fellowship that has facilitated this study. The comments of three anonymous reviewers and the editorial process improved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Krzemińska, U., Morales, H.E., Greening, C. et al. Population mitogenomics provides insights into evolutionary history, source of invasions and diversifying selection in the House Crow (Corvus splendens) . Heredity 120, 296–309 (2018). https://doi.org/10.1038/s41437-017-0020-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-017-0020-7
This article is cited by
-
The genetic drivers for the successful invasive potential of a generalist bird, the House crow
Biological Invasions (2022)
-
Culicidae evolutionary history focusing on the Culicinae subfamily based on mitochondrial phylogenomics
Scientific Reports (2020)
