
Abstract
Purpose
To assess the efficacy of tasimelteon to improve sleep in Smith–Magenis syndrome (SMS).
Methods
A 9-week, double-blind, randomized, two-period crossover study was conducted at four US clinical centers. Genetically confirmed patients with SMS, aged 3 to 39, with sleep complaints participated in the study. Patients were assigned to treatment with tasimelteon or placebo in a 4-week crossover study with a 1-week washout between treatments. Eligible patients participated in an open-label study and were followed for >3 months.
Results
Improvement of sleep quality (DDSQ50) and total sleep time (DDTST50) on the worst 50% of nights were primary endpoints. Secondary measures included actigraphy and behavioral parameters. Over three years, 52 patients were screened, and 25 patients completed the randomized portion of the study. DDSQ50 significantly improved over placebo (0.4, p = 0.0139), and DDTST50 also improved (18.5 minutes, p = 0.0556). Average sleep quality (0.3, p = 0.0155) and actigraphy-based total sleep time (21.1 minutes, p = 0.0134) improved significantly, consistent with the primary outcomes. Patients treated for ≥90 days in the open-label study showed persistent efficacy. Adverse events were similar between placebo and tasimelteon.
Conclusion
Tasimelteon safely and effectively improved sleep in SMS.
Similar content being viewed by others


INTRODUCTION
Smith–Magenis syndrome (SMS; OMIM 182290) is a rare genetic disorder that results from an interstitial deletion of 17p11.2 and, in rare cases, from a retinoic acid induced 1 (RAI1) gene variant [1]. The prevalence is estimated to be 1/15,000–25,000 [2, 3]. Recently, advancements in genetic testing and educational awareness of SMS have led to increased diagnosis of the syndrome among patients with neurodevelopment deficits [3]. Individuals with SMS present with a distinct pattern of mild to moderate intellectual disability, delayed speech and language skills, distinctive craniofacial and skeletal abnormalities, behavioral disturbances, and, almost uniformly, significant sleep disturbances [4]. Currently, the prevailing theory is that there is an underlying circadian pathophysiology causing sleep disturbances in these patients, as they exhibit low overall melatonin concentrations and abnormal timing of peak plasma melatonin concentrations. This abnormal inverted circadian rhythm is estimated to occur in 95% of individuals with SMS [5, 6].
RAI1 is a dosage-sensitive gene expressed in many tissues and highly conserved among species. Many studies have demonstrated that RAI1 and its homologs act as a transcriptional factor implicated in embryonic neurodevelopment and neuronal differentiation, as well as behavioral functions and circadian activity. Patients with RAI1 pathogenic variants show some phenotypic differences when compared to those carrying the typical deletion [7]; however, haploinsufficiency of RAI1 is the primary cause of the neurobehavioral and metabolic phenotype in SMS.
The 17p11.2 deletion encompasses RAI1, leading to haploinsufficiency, which is considered the primary cause for most features of SMS, including dysregulation of the molecular clock via its effect on CLOCK expression. ChIP-Chip and reporter studies suggest that RAI1 binds, directly or in a complex, to the first intron of CLOCK, enhancing transcriptional activity; thus, reduced expression of RAI1 results in reduced CLOCK expression in both animal models and SMS patient-derived cells [8]. These data suggest that treatment with a circadian regulator can, in part, correct the deficiencies caused by RAI1 abnormalities, providing further evidence of RAI1 interaction with the molecular clock and the impact on circadian rhythm.
The severe sleep disorder seen in this population causes significant disruption in the lives of individuals with SMS and their families. Sleep for these patients is characterized by difficulty sleeping at night and resultant excessive daytime sleepiness [9,10,11,12,13]. Individuals with SMS have decreased total night sleep, lower sleep efficiency, earlier sleep onset, final sleep offset, and increased waking after sleep onset compared to healthy individuals of the same age [14]. During these nighttime awakenings, individuals with SMS can pose a significant danger to themselves and disrupt the sleep of their family members. Such challenges that families of individuals with SMS face are well documented, and discussed by the advocacy group Parents & Researchers Interested in Smith–Magenis Syndrome (PRISMS) in their Medical Management Guidelines for an Individual Diagnosed with SMS [15].
Prior to the approval of tasimelteon for the treatment of nighttime sleep disturbances in Smith–Magenis Syndrome, there were no FDA-approved treatments for the disrupted sleep patterns associated with SMS. Multiple unapproved treatments are used with limited efficacy and with significant side effects, including hypnotic drugs, antidepressants, antipsychotics, sleep aids, mood stabilizers, α-2 agonists, and benzodiazepines [16]. Previous treatments for the sleep disorder in SMS include melatonin, oral β-1-adrenergic antagonists, and acebutolol with melatonin [17]. The support for this treatment regimen comes from early anecdotal reports and uncontrolled studies. In addition to medications, other methods caregivers use for managing SMS-related behaviors include using locks on doors and safety sleepers.
Tasimelteon is a melatonin receptor agonist that demonstrates high affinity for both the human melatonin MT1 and MT2 receptors. By acting upon the MT1 and MT2 receptors, tasimelteon acts by entraining circadian sleep phase timing and has been shown to improve nighttime sleep as well as daytime sleepiness and functioning [18,19,20]. Tasimelteon is approved by the US FDA and the EMA for the treatment of non-24-hour sleep–wake disorder (non-24) and by the US FDA for the treatment of nighttime sleep disturbances in Smith–Magenis syndrome (SMS). The SET and RESET studies that supported the approval of tasimelteon for non-24 demonstrated tasimelteon’s circadian entraining properties and its ability to improve sleep parameters [19].
Given the suspected circadian nature underlying the sleep disruption in SMS, we hypothesized that tasimelteon could ameliorate this disruption and would lead to improved sleep in individuals with SMS.
MATERIALS AND METHODS
Study design
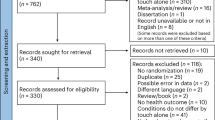
This study was a placebo-controlled, double-blind, randomized, two-period crossover study followed by an optional open-label extension phase to evaluate the safety and efficacy of tasimelteon in improving the sleep disorder in SMS. As shown in Fig. 1, the study consisted of three phases: a screening phase, a treatment phase, and an open-label extension phase for those who wished to continue treatment. During the treatment phase, patients who did not meet criteria for randomization into the crossover portion were given the option to enroll into open-label. During the crossover portion of the treatment phase, both study patients and the medical professionals interacting with patients were unaware of treatment group assignment. Because SMS is a relatively uncommon syndrome, the population eligible to enroll into this study was limited. A crossover design was chosen to accommodate the small population size available and to increase statistical power.

(a) CONSORT patient flow diagram. (b) The study consisted of a randomized, placebo-controlled, two-phase crossover treatment arm and a parallel open-label treatment arm for patients who did not qualify for the randomization treatment period. Both the randomization and open-label treatment arms were followed by an open-label extension phase.
To be eligible for the study, candidates must have had a confirmed clinical and genetic diagnosis of SMS with chromosomal microarray or targeted sequencing (see Table 1), between the ages of 3 and 65 years of age and had a recent history of sleep disturbances. Patients were also required to have a caregiver able to complete outpatient assessments and to be willing and able to comply with study requirements.
Those who fulfilled these criteria entered the screening phase. Patients who met all initial eligibility criteria at screening began washing out of prohibited medications. Those unwilling or unable to follow the medication restrictions including the washout from use of a prohibited medication were excluded. Prohibited concomitant medications included any medication known to cause sedation or stimulation, dietary supplements and other preparations containing melatonin, and melatonin agonists. Candidates who demonstrated fragmented sleeping and impaired sleep quality during the screening phase were eligible to enter the study.
The study was conducted at four sites in the United States. To reduce the burden on patients and their families, for most visits, patients were permitted to either report to the site closest to them or have a trained nurse conduct the visit in the patient’s home.
During the crossover portion of the study, patients were randomly assigned to one of two treatment sequences. Those assigned to treatment sequence A were dosed with tasimelteon for four weeks, followed by a washout period of one week. Following the washout period, those in treatment sequence A received four weeks of treatment with placebo. Those assigned to treatment sequence B received placebo for four weeks, followed by a washout period of one week. Following the washout period, those in treatment sequence B received four weeks of treatment with tasimelteon.
Treatment
Two formulations of tasimelteon were used in this study. A capsule (20 mg) was provided for adult patients and an oral suspension administered by 3 ml or 5 ml syringe (4 mg/ml) was provided for pediatric patients. A weight appropriate dosing schedule was developed for the oral suspension in a previous study and administered as ≤28 kg: 0.7 mg/kg or >28 kg: 20 mg. Treatment was administered as a capsule or oral suspension once daily, one hour before target bedtime (±30 minutes). Placebo and tasimelteon were indistinguishable regardless of formulation.
Statistical analysis
The primary endpoints were the average of 50% worst daily diary sleep quality (DDSQ50) (change from baseline) and the average of the 50% worst daily diary total sleep time (DDTST50). These primary endpoints looked at the worst half of nights for sleep quality (DDSQ50) and total sleep time (DDTST50). These data were collected through a daily diary that the patients’ caregivers completed on provided forms. These endpoints were developed through a preliminary, observational study in which the sleep patterns of SMS patients were observed. During the observational study it was observed that patients had highly variable sleep patterns with nights of good and poor quality sleep and of long and short total sleep time. Thus, to better quantify the effect of tasimelteon in this population, the DDSQ50 and DDTST50 were chosen as primary endpoints. The average of 50% worst nighttime sleep quality (DDSQ50) was calculated as the average of the first half of data, sorted from worst to best, if the total number of data points was even, or the first half +1 of data if the total number of data points was odd. For DDTST50 the same approach was utilized. These endpoints were analyzed in the intent-to-treat (ITT) population by comparing the effect of tasimelteon and placebo. P values were calculated using a mixed effects model that included the fixed, categorical effects of treatment, period, and sequence of treatment.
Secondary endpoints that were analyzed included the average daily diary sleep quality (DDSQ), daily diary total sleep time (DDTST), 50% worst daily number of nighttime awakenings, 50% worst latency to sleep, daily number of nighttime awakenings, daily total amount of nighttime sleep, and latency to sleep, all by post sleep questionnaire (PSQ) and actigraphy. Additional secondary endpoints included the average 50% worst daily length of nighttime awakenings, 50% worst daily total amount of nighttime sleep, and daily length of nighttime awakenings measured by actigraphy. Finally, changes in clinical impression were determined as Clinical Global Impression Severity and Change (CGI-S, CGI-C) at each visit, and mixed model analysis was used to analyze the treatment differences using the Aberrant Behavior Checklist (ABC) [21, 22].
Data from the open-label treatment arm and open-label extension are also presented below. The same measures were collected as in the randomization arm and are used to provide additional insight into the persistence of efficacy of tasimelteon.
Clinical genetic testing
Confirmation of Smith–Magenis diagnosis was determined for each participant by clinical chromosomal microarray assay (CMA) or targeted RAI1 gene sequencing. Participants had a confirmed heterozygous deletion (of chromosome 17p11.2) inclusive of RAI1 or an RAI1 pathogenic variant resulting in haploinsufficiency (Table 1). CMA identified a copy-number loss of chromosome band encompassing the RAI1 gene in the Smith–Magenis, as depicted in Table 1. Of the 38 patients, 37 patients had hemizygosity of RAI1 due to 17p11.2, while one patient had a RAI1 variant (RAI1 stop-gain). Moreover two patients' deletions were larger in size and encompassed deletion of CMT1A (Table 1 bold).
RESULTS
Potential patients were identified from SMS registries operated by Vanda; advocacy groups including PRISMS, Inc.; and through phone and web-based patient outreach campaigns. Patient recruitment for the crossover portion of the study began in 2015 and lasted for approximately 3 years. Figure 1a shows the flow diagram of patients for the study, with 52 people screened as of the data lock point on 3 December 2018 and 46 people completing the screening phase. A total of 26 people were enrolled to the treatment phase of the study, 25 (96.2%) who were randomized completed the treatment phase, and only 1 person (3.8%) withdrew from the treatment phase. Following completion of the treatment phase, randomized patients were offered enrollment into the open-label extension (OLE) phase; however, 19 of the 52 screened study patients (36.5%) entered directly into the OLE phase. Five patients were initially assigned to the open-label phase and then subsequently rescreened into the randomization phase (an additional subgroup analysis excluding these patients is provided below). Supplementary Table 1 summarizes the demographic characteristics for all randomized patients. Overall, demographic and other baseline characteristics were evenly distributed between both treatment arms.
Results from the ITT population were analyzed for all primary and secondary efficacy endpoints. Both primary endpoints, average of 50% worst DDSQ (DDSQ50) and average of 50% worst DDTST (DDTST50), favored tasimelteon over placebo, with DDSQ50 demonstrating a statistically significant improvement over placebo (Table 2). Treatment with tasimelteon showed a difference of 0.4 increase in average sleep quality on the worst 50% of nights (tasimelteon = 2.8, placebo = 2.4, p = 0.0139). Total sleep time on the worst 50% of nights resulted in a difference of 18.5 minutes increase for tasimelteon compared to placebo (tasimelteon = 419.3 minutes, placebo = 400.9 minutes, p = 0.0556).
Secondary endpoints showed both subjective and objective evidence of tasimelteon treatment improving sleep related symptoms. These results complemented and extended the conclusions from the primary endpoints showing improvement in overall sleep quality (tasimelteon = 0.6, placebo = 0.2, p = 0.0155) and total sleep time as determined by diary (tasimelteon = 40.9, placebo = 19.8, p = 0.0134). Further, actigraphy-based measurement of total sleep time also showed improvement (tasimelteon = 20.2, placebo = 1.9, p = 0.0218). Secondary efficacy endpoints are summarized in Table 3.
Subgroup analysis was performed after removing the five patients who were randomized after having enrolled in open-label treatment (Table 4). The results of this subgroup analysis were similar to the full ITT. Both primary endpoints (DDSQ50 and DDTST50) demonstrated statistical and clinical significance, as did the overall average (DDSQ and DDTST).
The open-label extension study showed consistent improvement in sleep with tasimelteon treatment in DDSQ50, DDSQ, DDTST50, and DDTST (Supplementary Table 2). The magnitude of improvement in these sleep related symptoms was equivalent across treatment phases, providing further evidence of the treatment effect. Further, objective measures of sleep, i.e., actigraphy, showed that total sleep time was similar across the double-blind and open-label periods of treatment. Interestingly, a larger improvement in behavioral symptoms was observed in patients treated for longer than 90 days as measured by the ABC (Supplementary Table 25).
Safety analyses were conducted for both the treatment phase and the open-label extension. Overall, tasimelteon was safe and well tolerated. For the treatment phase, 7 (26.9%) patients experienced a treatment emergent adverse events (TEAE) while on placebo, and 6 (24.0%) patients experienced a TEAE while on blinded tasimelteon. Ten (52.6%) patients reported a TEAE while on open-label tasimelteon. One (4.0%) patient on blinded tasimelteon and one (5.3%) patient on open-label tasimelteon each experienced a TEAE that led to a temporary disruption of the study drug; no patients experienced any TEAEs that led to study drug being withdrawn. There were no serious adverse events or deaths during the treatment phase. There were no clinically meaningful changes in mean chemistry, hematology, or urinalysis values over time. In general, the adverse events experienced were common for the study population and similar between placebo and tasimelteon for the treatment phase.
DISCUSSION
Tasimelteon provided a significant improvement in sleep in patients with SMS treated for 4 weeks in a double-blind, placebo-controlled, crossover study. This improvement was observed in the primary endpoint of 50% worst nights of sleep quality (DDSQ50) (difference = 0.4, p = 0.0139). The second primary endpoint did not reach statistical significance but showed an average improvement of 18.5 minutes of sleep (p value = 0.0556) on the worst 50% of nights (DDTST50). In addition to the improvement demonstrated on the primary endpoints, tasimelteon improved additional subjective and objective measures that characterize both the quality and length of sleep. This is particularly noteworthy given that sleep disturbances in those with SMS have been difficult to improve; in fact, in a study assessing the use of prolonged-release melatonin to improve insomnia in children with an autism spectrum disorder and/or neurodevelopmental disorder, results showed a greater improvement in Total Sleep Time (TST) when SMS patients were excluded from analysis [23].
In addition to the effects observed in the crossover portion of the study (4 weeks of treatment), there was an OLE phase into which patients from the randomization and open-label treatment periods could enter. During the OLE phase, tasimelteon continued to show improved sleep by both objective and subjective measures. Interestingly, while the ABC checklist that quantifies behavioral symptoms in SMS did not show statistically significant improvement in the 4-week randomization phase, a large improvement emerged in the OLE phase when the length of treatment was extended for 3 months or longer. This suggests that sleep improvements take additional time to engender behavioral improvements and is consistent with previous findings [23].
One limitation of this study is the relatively small sample size due to the rarity of the disorder and the difficulty of patients to participate in a controlled study, which limits the types of analyses and evaluations of the efficacy of the drug. Another limitation of the study is the duration of the washout period of one week; however, the washout was limited to one week to reduce the amount of time patients were without treatment. Due to logistics, and difficulty bringing in patients for the serum melatonin measurements, melatonin was not measured in this current study. That is one limitation that could be addressed in future studies either via serum melatonin measurement or via salivary dim light melatonin onset (DLMO) tests.
The results of this study are consistent with the putative mechanism of action of tasimelteon as a circadian regulator in non-24 and jet-lag disorder [18, 19]. The underlying genetic etiology of SMS, haploinsufficiency of RAI1 due to chromosome 17p11,2 deletion or pathogenic variant in RAI1, causes circadian dysfunction in patients, leading to a myriad of symptoms, most critically, sleep disruption. In this study, we provide evidence that tasimelteon can safely provide persistent improvement of the sleep disturbances associated with SMS and may ultimately improve the quality of life for patients and their families. Additional longitudinal real world studies should be conducted to evaluate the benefits of tasimelteon in improving both sleep and behavioral symptoms of patients with SMS.
References
Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith–Magenis syndrome. Nat Genet. 2003;33:466–8.
Greenberg F, Guzzetta V, Montes de Oca-Luna R, Magenis RE, Smith AC, Richter SF, et al. Molecular analysis of the Smith–Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2). Am J Hum Genet. 1991;49:1207–18.
Smith AC, Magenis RE, Elsea SH. Overview of Smith–Magenis syndrome. J Assoc Genet Technol. 2005;31:163–7.
Potocki L, Shaw CJ, Stankiewicz P, Lupski JR. Variability in clinical phenotype despite common chromosomal deletion in Smith–Magenis syndrome [del(17)(p11.2p11.2)]. Genet Med. 2003;5:430–4.
Boone PM, Reiter RJ, Glaze DG, Tan DX, Lupski JR, Potocki L. Abnormal circadian rhythm of melatonin in Smith–Magenis syndrome patients with RAI1 point mutations. Am J Med Genet A. 2011;155A:2024–7.
Spruyt K, Braam W, Smits M, Curfs LM. Sleep complaints and the 24-h melatonin level in individuals with Smith–Magenis syndrome: assessment for effective intervention. CNS Neurosci Ther. 2016;22:928–35. https://doi.org/10.1111/cns.12653.
Girirajan S, Vlangos CN, Szomju BB, Edelman E, Trevors CD, Dupuis L, et al. Genotype-phenotype correlation in Smith–Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet Med. 2006;8:417–27.
Williams SR, Zies D, Mullegama SV, Grotewiel MS, Elsea SH. Smith–Magenis syndrome results in disruption of CLOCK gene transcription and reveals an integral role for RAI1 in the maintenance of circadian rhythmicity. Am J Hum Genet. 2012;90:941–9.
Greenberg F, Lewis RA, Potocki L, Glaze D, Parke J, Killian J, et al. Multi-disciplinary clinical study of Smith–Magenis syndrome (deletion 17p11.2). Am J Med Genet. 1996;62:247–54.
Smith AC, Dykens E, Greenberg F. Sleep disturbance in Smith–Magenis syndrome (del 17 p11.2). Am J Med Genet. 1998;81:186–91.
Potocki L, Glaze D, Tan DX, Park SS, Kashork CD, Shaffer LG, et al. Circadian rhythm abnormalities of melatonin in Smith–Magenis syndrome. J Med Genet. 2000;37:428–33.
De Leersnyder H, de Blois M-C, Claustrat B, Romana S, Albrecht U, Von Kleist-Retzow JC, et al. Inversion of the circadian rhythm of melatonin in the Smith–Magenis syndrome. J Pediatr. 2001;139:111–6.
Smith ACM, Duncan WC. Smith–Magenis syndrome—a developmental disorder with circadian dysfunction. In: Genetics of developmental disabilities. Boca Raton, FL: Taylor & Francis; 2005.
Smith ACM, Morse RS, Introne W, Duncan WC. Twenty-four-hour motor activity and body temperature patterns suggest altered central circadian timekeeping in Smith–Magenis syndrome, a neurodevelopmental disorder. Am J Med Genet Part A. 2019;179:224–36.
Smith ACM, Boyd KE, Brennan C, et al. Medical management guidelines for an individual diagnosed with SMS. PRISMS. Retrieved from https://www.prisms.org/wpcontent/uploads/pdf/mmg/PRISMS_Medical_Management_Guidelines2018.pdf. 2018.
Laje G, Morse R, Richter W, Ball J, Pao M, Smith ACM. Autism spectrum features in Smith–Magenis syndrome. Am J Med Genet Part C Semin Med Genet. 2010;154:456–62.
Smith ACM, Boyd KE, Elsea SH, et al. Smith–Magenis syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews. Seattle: University of Washington; 1993.
Rajaratnam SM, Polymeropoulos MH, Fisher DM, Roth T, Scott C, Birznieks G, et al. Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials. Lancet. 2009;373:482–91.
Lockley SW, Dressman MA, Licamele L, Xiao C, Fisher DM, Flynn-Evans EE, et al. Tasimelteon for non-24-hour sleep-wake disorder in totally blind people (SET and RESET): two multicentre, randomised, double-masked, placebo-controlled phase 3 trials. Lancet. 2015;386:1754–64.
Polymeropoulos CM, Mohrman MA, Keefe MS, Brzezynski JL, Wang J, Prokosch LS, et al. Efficacy of tasimelteon (HETLIOZ®) in the treatment of jet lag disorder evaluated in an 8-h phase advance model; a multicenter, randomized, double-blind, placebo-controlled trial. Front Neurol. 2020;11:1–10.
Busner J, Targum SD. The Clinical Global Impressions Scale: applying a research tool in clinical practice. Psychiatry. 2007;4:28–37.
Aman MG, Singh NN, Stewart AW, Field CJ. The aberrant behavior checklist: a behavior rating scale for the assessment of treatment effects. Am J Ment Defic. 1985;89:485–91.
Schroder CM, Malow BA, Maras A, Melmed RD, Findling RL, Breddy J, et al. Pediatric prolonged-release melatonin for sleep in children with autism spectrum disorder: impact on child behavior and caregiver’s quality of life. J Autism Dev Disord. 2019;49:3218–30.
Acknowledgements
This work was supported by Vanda Pharmaceuticals Inc. We are very grateful to the participants and their families for their efforts and participation and to the investigators in the study. We are also very grateful to PRISMS.
Author information
Authors and Affiliations
Contributions
Conceptualization: C.M.P., G.B., M.P. Data curation: C.X., C.M.P., E.L.C., K.K., M.M.G., J.B., S.P.S. Formal analysis: C.X., E.L.C., S.P.S., J.B., C.M.P. Funding acquisition: C.M.P., M.H.P. Investigation: C.M.P., G.B., M.H.P., S.H.E. Resources: C.P., M.H.P. Software: C.X., S.P.S. Visualization: C.X., E.C., K.K. Writing—original draft: E.L.C., K.K., M.M.G., J.B., S.P.S., J.B., C.P. Writing—review & editing: S.P.S.
Corresponding author
Ethics declarations
Competing interests
C.M.P., J.B., E.L.C., M.A.F., M.M.G., K.K., S.P.S., C.X., G.B., M.H.P. are employees of Vanda Pharmaceuticals. M.H.P. is Chief Executive Officer of Vanda Pharmaceuticals. S.H.E. declares no competing interests.
Ethics statement
Patients or their legal guardian signed informed consent forms before any study assessments, including screening phase assessments. The study was approved by Copernicus Group Independent Review Board and by Western Institutional Review Board (WIRB).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data Sharing Statement: Data available upon request.
IRB approval status: Reviewed and approved by the central IRB is WIRB-Copernicus Group (WCG) IRB tracking number 420150480. Three of the four study sites (Site 101/Emsellem, Site 102/Norman, and Site 104/Marriott) are under the central IRB. The fourth site (Site 103/Elsea) is under a local IRB, Western Institutional Review Board (WIRB).
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Polymeropoulos, C.M., Brooks, J., Czeisler, E.L. et al. Tasimelteon safely and effectively improves sleep in Smith–Magenis syndrome: a double-blind randomized trial followed by an open-label extension. Genet Med 23, 2426–2432 (2021). https://doi.org/10.1038/s41436-021-01282-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01282-y
This article is cited by
-
Pharmacologic Approaches to Insomnia and Other Sleep Disorders in Children
Current Treatment Options in Neurology (2022)
