
Abstract
Purpose
DYRK1A syndrome is among the most frequent monogenic forms of intellectual disability (ID). We refined the molecular and clinical description of this disorder and developed tools to improve interpretation of missense variants, which remains a major challenge in human genetics.
Methods
We reported clinical and molecular data for 50 individuals with ID harboring DYRK1A variants and developed (1) a specific DYRK1A clinical score; (2) amino acid conservation data generated from 100 DYRK1A sequences across different taxa; (3) in vitro overexpression assays to study level, cellular localization, and kinase activity of DYRK1A mutant proteins; and (4) a specific blood DNA methylation signature.
Results
This integrative approach was successful to reclassify several variants as pathogenic. However, we questioned the involvement of some others, such as p.Thr588Asn, still reported as likely pathogenic, and showed it does not cause an obvious phenotype in mice.
Conclusion
Our study demonstrated the need for caution when interpreting variants in DYRK1A, even those occurring de novo. The tools developed will be useful to interpret accurately the variants identified in the future in this gene.
Graphic abstract

Similar content being viewed by others

INTRODUCTION
Intellectual disability (ID) and autism spectrum disorder (ASD) are two highly heterogeneous groups of neurodevelopmental disorders (NDDs) with substantial genetic contributions that overlap strongly both at the clinical and genetic levels. More than 1,000 genes have been implicated in monogenic forms of NDD, with an important contribution of autosomal dominant forms caused by de novo variants [1]. One of these genes, DYRK1A (dual-specificity tyrosine phosphorylation-regulated kinase 1A) [2], located on chromosome 21, is among the genes the most frequently mutated in individuals with ID [1].
The first DYRK1A disruptions were identified in individuals with intrauterine growth restriction (IUGR), primary microcephaly, and epilepsy [3]. A few years later, the first frameshift variant was described in a patient with similar features [4]. The clinical spectrum associated with DYRK1A pathogenic variants (MRD7; mental retardation 7 in OMIM) was further refined with the publication of additional patients, presenting suggestive facial dysmorphism, severe speech impairment, and feeding difficulty, while epilepsy and prenatal microcephaly were not always present [5,6,7,8,9]. Pathogenic variants were also identified in cohorts of individuals with ASD [10], but all have ID [11]. The DYRK1A gene encodes a dual tyrosine–serine/threonine (Tyr-Ser/Thr) kinase composed of a central catalytic domain including Tyrosine 321, involved in DYRK1A activation by autophosphorylation [12], two nuclear localization signal sequences (NLS), and additional functional domains. DYRK1A is ubiquitously expressed during embryonic development and in adult tissues. Its location is both cytoplasmic and nuclear and varies by cell type and stage of development [13]. By the number and diversity of its proposed protein targets, DYRK1A regulates numerous cellular functions (reviews [14, 15]), among them the MAPT (Tau) protein phosphorylated by DYRK1A on its Thr212 position [16].
High-throughput sequencing (HTS) has revolutionized the identification of genetic variants for diagnostic applications but a major challenge remains in the interpretation of the vast number of variants, especially for highly heterogeneous disease such as ID. A combination of genetic, clinical, and functional approaches, summarized by the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP), are commonly used to interpret these variants [17]. A significant proportion of the variants, especially the missense variants, remain classified as variants of unknown significance (VUS, according to ACMG/AMP) after the primary analysis. For autosomal dominant diseases with complete penetrance such as DYRK1A syndrome, the de novo occurrence of a variant is a strong but not absolute argument for pathogenicity, and the parents’ genotype is not always available. Clinical observations are useful but could also lead to misinterpretation, as numerous ID-associated manifestations are unspecific. Many tools have been developed to predict in silico the pathogenicity of missense variants but they remain predictions and protein-specific functional tests are useful to confirm variants' effects. DNA methylation (DNAm) is also a powerful tool to test variant pathogenicity in disorders associated with epigenetic regulatory genes. We discovered that pathogenic variants in these genes can exhibit disorder-specific DNAm signatures comprised of consistent, multilocus DNAm alterations in peripheral blood, useful for classifying variants in these genes as pathogenic or benign [18,19,20,21]. DYRK1A has numerous targets, and while it is not well-described as an epigenetic regulator, it has been shown to phosphorylate histone H3 and histone acetyltransferase proteins [22, 23]. Therefore, we hypothesized that pathogenic variants in DYRK1A might generate a specific DNAm signature in blood.
We reviewed the clinical signs present in 34 never-reported individuals who carried deletions or clearly pathogenic variants in DYRK1A to refine the clinical spectrum of DYRK1A syndrome. Based on these data, we developed a score to help to recognize affected individuals and to interpret variants (reverse phenotyping). In parallel, we developed in silico and in vitro approaches to assess variant effects on DYRK1A function. Finally, we defined a DNAm signature specific to DYRK1A syndrome in patient blood. We used this multifaceted approach to interpret 17 variants identified in patients with ID/NDD and demonstrated its efficiency in optimizing accurate interpretation of variants.
MATERIALS AND METHODS
Patients and molecular analysis
Variants in DYRK1A (NM_001396.4) were identified during routine genetic analyses in individuals referred to clinical genetic services for intellectual disability in France, Denmark, and Switzerland using the following: comparative genomic hybridization (CGH) array, Sanger sequencing of DYRK1A coding regions, targeted next-generation sequencing of ID genes (TES) [24,25,26], trio or simplex clinical exome sequencing (CES) or exome sequencing (ES), and were confirmed by an additional method. Combined Annotation Dependent Depletion (CADD) and Nnsplice were used to predict the effect of missense and splice variants respectively. Fibroblasts were established from skin biopsies for individuals 1, 11, 22, 24; Bronicki 9; and Bronicki 10 and cultivated as previously described [27]. Paxgene blood samples were collected for individuals 9, 18, 19, and 30. Messenger RNA (mRNA) extraction, RNA sequencing, reverse-transcription polymerase chain reaction (RT-PCR) or quantitative PCR (qPCR) (primers available on request) were performed as previously described [28].
Phenotypic analysis and clinical scoring
Clinical information and photographs were provided by the referring clinicians for the 44 individuals reported here (Table S1) as well as for six previously published individuals (Bronicki 2, 3, 8, 9, and 10, and Ruaud 2) [6, 7] (n = 50 individuals in total). Based on the most frequent signs and the morphometric characteristics presented by the 34 individuals with truncating variants in DYRK1A, a clinical score out of 20 was established (DYRK1A_I, n = 21 individuals with photographs available, Table S2). Clinical scores were calculated for this initial cohort plus a second cohort (replication cohort, DYRK1A_R, n = 13) with individuals already described in previous publications [5,6,7] by experienced clinical geneticists, as well as for individuals with other frequent monogenic forms of ID, caused by pathogenic variants in DDX3X (n = 5), ANKRD11 (n = 5), ARID1B (n = 8), KMT2A (n = 6), MED13L (n = 5), SHANK3 (n = 6), or TCF4 (n = 6).
Definition of sets of missense variants and conservation analysis
To evaluate which tools are pertinent to predict effect of missense variants on the DYRK1A protein, we used different sets of variants: (1) variants presumed to be benign (negative N-set, n = 115) including missense variants reported benign in ClinVar plus variants found more than once in gnomAD (November 2019 release), (2) variants reported as pathogenic/likely pathogenic in ClinVar (positive P-set, n = 16), and (3) other variants reported here, in literature, or as VUS in ClinVar (test T-set, n = 44)(Table S3). One hundred twenty-three orthologous sequences of human DYRK1A were extracted from the OrthoInspector database version 3.0. and a multiple sequence alignment (MSA) was constructed using the Clustal Omega software. The MSA was then manually refined to correct local alignment errors using the Jalview MSA editor [29]. The refined MSA was used as input to the PROBE software [30] to identify conserved regions in the sequences. The sequences in the MSA were divided into five separate clades: vertebrates, metazoans, protists, plants, and fungi.
In vitro analysis of variant effect on DYRK1A protein
DYRK1A expression plasmids (addgene #101770, Huen lab) were generated from the pMH-SFB-DYRK1A vector containing the N-terminal FLAG-tagged human DYRK1A complementary DNA (cDNA) sequence (NM_001396.4). Variant sequences were obtained by site-directed mutagenesis as described [31]. Antibodies used are listed in Supplementary Methods. HeLa, HEK293, and COS1 cells were maintained and transfected for 24 hours with DYRK1A plasmids (for immunoflurorescence) or DYRK1A plasmids plus pEGFP-N1 plasmid (for western blot) as previously done [31, 32]. For autophosphorylation analysis and DCFA7 (WDR68) interaction, proteins were extracted from transfected HEK293 cells and immunoprecipitated with anti-FLAG antibody as described [31]. Phosphorylated Tyr321 DYRK1A was visualized as described [33] and normalized by the level of total DYRK1A protein. Kinase activity was investigated by cotransfecting DYRK1A and MAPT plasmids (MAPT_OHu28029C_pcDNA3.1(+)-C-HA, geneScript) in HEK293 cells, adapted from [34]. The level of DYRK1A, MAPT, and pMAPT (Thr212) proteins were normalized with GAPDH.
DNA methylation signature
Methylation analysis was performed using blood DNA from individuals with DYRK1A loss-of-function (LoF) variants (n = 16), split into signature discovery (n = 10) and validation (n = 6) cohorts, based primarily on whether age at time of blood collection was available, and age- and sex-matched neurotypical controls (n = 24). Whole-blood DNA samples were prepared, hybridized to the Illumina Infinium Human MethylationEPIC BeadChip, and analyzed as previously described [20]. A total of n = 774,590 probes were analyzed for differential methylation. Standard quality control metrics showed good data quality for all samples except individual 20. Briefly, limma regression with covariates age, sex, and five predicted blood cell types identified a DNAm signature with a Benjamini–Hochberg adjusted p value <0.05 and 10% methylation difference. Next, we developed a support vector machine (SVM) model with linear kernel trained on including nonredundant CpG sites [20] using the methylation values for the discovery cases versus controls. The model generated scores ranging between 0 and 1 for tested samples, classifying samples as positive (score > 0.5) or negative (score < 0.5). Additional neurotypical controls (n = 94) and DYRK1A LoF validation samples (n = 6) were scored to test model specificity and sensitivity respectively, and samples with pathogenic KMT2A (n = 8) and ARID1B (n = 4) variants and DYRK1A missense and distal frameshift (n = 11) variants were tested.
RESULTS
Identification of genetic variants in DYRK1A in individuals with ID
We collected molecular and clinical information from 50 individuals with ID (44 never reported and six previously reported [6, 7]) carrying a variant in DYRK1A identified in clinical and diagnostic laboratories: structural variants deleting or interrupting DYRK1A and recurrent or novel nonsense, frameshift, splice, and missense variants (Table 1, Figure S1). When blood or fibroblast samples were available, we characterized the consequences of these variants on DYRK1A mRNA by RNA sequencing and RT-qPCR (Figure S2, Supplementary Text). For one variant, c.1978del, occurring in the last exon of the gene (individual 18), the mutant transcripts escape to nonsense-mediated mRNA decay (NMD) and result in a truncated protein p.Ser660fs (or p.Ser660Profs*43) retaining its entire kinase domain (Figure S2F). The variants occurred de novo in most of the cases (42/50), one was inherited from a mosaic father, and parental DNA was not available for the remaining seven cases.
Clinical manifestations in individuals with pathogenic variants in DYRK1A and definition of a clinical score
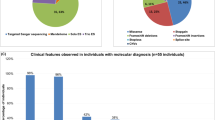
We reviewed the clinical manifestations of the patients with truncating variants, except p.(Ser660fs) (Table S1, Supplementary Text). Recurrent features include, consistently with what was reported [5, 6, 8, 11], moderate to severe ID, prenatal or postnatal progressive microcephaly, major speech impairment, feeding difficulties, seizures and especially history of febrile seizures, autistic traits and anxiety, delayed gross motor development with unstable gait, brain magnetic resonance imaging (MRI) abnormalities including dilated ventricles and corpus callosum hypoplasia, and recurrent facial features (Fig. 1a, Figure S3). We found genital abnormalies as reported [35] but no obvious renal anomalies. We noted the importance of skin manifestations and especially atopic dermatitis. We used recurrent features to establish a DYRK1A clinical score (CSDYRK1A) on a 20-point scale (Fig. 1a), which aims to reflect specificity rather than severity of the phenotype. High scores, ranging from 13 to 18.5 (mean = 15.5), were obtained for the individuals having a pathogenic variant in DYRK1A described here (DYRK1A_I) or previously (DYRK1A_R) (Table S2, Fig. 1b). The threshold of CSDYRK1A ≥13 appears to be discriminant between individuals with LoF variants in DYRK1A (all ≥ 13) and individuals suffering from another form of ID (all < 13) and is considered as highly suggestive (13 > CSDYRK1A > 10: intermediate; CSDYRK1A < 10: poorly evocative). A clinical score of 15 points without photograph is less discriminative (Figure S4).

(a) Clinical score out of 20 points established according to the most recurrent clinical features presented by patients (the weight assigned to each symptom being based on its recurrence): clinical symptoms are out of 15 points, while the facial appearance is out of 5 points. CA cerebral atrophy, CCA/H corpus callosum agenesis or hypoplasia, CeA cerebellar atrophy, EV enlarged ventricules. (b) Clinical scores calculated for individuals carrying pathogenic variants in DYRK1A reported here and for whom photographs were available (n = 21) (initial cohort, DYRK1A_I, scores 13–17.5 with a mean of 15.5), the previously published individuals (replication cohort, DYRK1A_R, scores 13.5–18.5, mean = 15.3) and the individuals affected with other frequent monogenic forms of intellectual disability (ID), associated to variants in ANKRD11, MED13L, DDX3X, ARID1B, SHANK3, TCF4, or KMT2A (scores 3–12.5, mean = 7). The clinical score for the individuals carrying missense or distal frameshift variants are indicated in yellow (test); the threshold of CSDYRK1A ≥13 appeared to be discriminant between individuals with loss-of-function (LoF) variants in DYRK1A (all ≥ 13) and individuals suffering from another form of ID (all < 13). A score above this threshold was therefore considered highly suggestive. We classified individuals with CSDYRK1A < 10 as poorly evocative and individuals with a CSDYRK1A between 10 and 13 as intermediate. Brown–Forsythe and Welsh analysis of variance (ANOVA) tests with Dunnett’s T 3 multiple comparisons test were performed. ns not significant; **p < 0.01; ***<p < 0.001; error bars represent SD.
In silico analysis of missense variant effects
We evaluated the discriminative power of the CADD score, commonly used in medical genetics [36] to interpret missense variants in DYRK1A. If a significant difference in the CADD score distribution is observed between the benign variants (N-set, see “Materials and Methods”) and those reported as pathogenic (P-set, see “Materials and Methods”) (p value <0.0001), a substantial proportion of the N-set variants still have a CADD score above thresholds (20 or 25) usually used to define pathogenicity (Figure S5A). This could be explained by the high degree of amino acid conservation of DYRK1A among vertebrates, and this could lead to overinterpretation of pathogenicity. We performed sequence alignment with orthologs from different taxon (Figure S6) and confirmed that using sequences from vertebrates only is not efficient to classify missense variants, as one third of the N-set variants affect amino acids conserved in all species (Figure S5B, V = 100%). Considering conservation parameters going beyond vertebrates appears more discriminant (13/16 P-set and 1/115 N-set variants are conserved in 100% of vertebrates, 90% of metazoans, and 80% of others) (Figure S5B).
In vitro characterization of consequences of missense variants on DYRK1A protein
To test the consequences of the missense variants in vitro, we overexpressed wild-type (WT) and mutant DYRK1A proteins in three different cell lines (HEK293, HeLa, COS1) including a truncating pathogenic variant Arg413fs and a benign missense variant from gnomAD, Ala341Ser. A significant and drastic decrease in DYRK1A protein level (Fig. 2a), due to a reduction of protein stability (Figure S7A), was observed for the truncating Arg413fs and missense Asp287Val, Ser311Phe, Arg467Gln, Gly168Asp, and Ile305Arg variants in each cell type. None of the variants affects DYRK1A interaction with DCAF7 (WDR68) (Figure S7B). To be active, DYRK1A has to undergo an autophosphorylation on Tyrosine 321 [12]. To measure the level of active DYRK1A protein, we detected phospho-DYRK1A (Tyr321) by immunoprecipitation followed by immunoblot using anti-phospho-HIPK2, as previously described [33] (Fig. 2b). We confirmed that the three variants previously tested (Asp287Val, Ser311Phe, and Arg467Gln) abolish autophosphorylation [33, 37], as do the Gly168Ap and Ile305Arg variants. The Ser324Arg DYRK1A variant showed only residual autophosphorylation. No effect on autophosphorylation was observed for Arg255Gln, Tyr462His, Gly486Asp, and Thr588Asn. No effect was detected either for the Glu366Asp amino acid change, but the analysis of patient’s blood mRNA showed that this variant (c.1098G>T) affects splicing leading to a deletion of 49 amino acids p.Ile318_Glu366del (Figure S2G). We used this strategy to test additional variants reported in databases and showed that Arg158His, affecting a highly conserved amino acid position but reported twice in gnomAD, does not affect DYRK1A protein. Ala277Pro, reported as pathogenic in ClinVar, as well as Gly171Arg, Leu241Pro and Pro290Arg, reported initially as VUS in ClinVar, affect both DYRK1A level and autophosphorylation (Figure S8A, B, Table S3, Figure S5B). None of the missense variants appears to affect DYRK1A cellular localization, contrary to Arg413fs variant or changes in NLS domains (Supplementary Text, Figure S8C). However, we observed an aggregation of DYRK1A proteins with the distal frameshift variant Ser660fs (Fig. 2c), preventing a correct measure of the protein and autophosphorylation levels.

(a) Level of variant DYRK1A proteins expressed in HeLa, HEK293, and COS cells transiently transfected with DYRK1A constructs. Protein levels were normalized on the level of GFP proteins (expressed from a cotransfected pEGFP plasmid). Quantifications were performed on a total of n ≥ 9 series of cells (n ≥ 3 Hela cells, n ≥ 3 HEK293, and n ≥ 3 COS cells) using ImageJ software. One-way analysis of variance (ANOVA) with multiple comparison test was performed to compare the level of variant DYRK1A proteins to the level of wild‐type DYRK1A protein (orange dashes), applying Bonferroni’s correction. ns not significant; *p < 0.05; **p < 0.01; ***p < 0.001; error bars represent SEM. Green, the variant from gnomAD; red, a truncating variant; gold, the variants tested in this study. (b) DYRK1A’s ability to autophosphorylate on Tyr321 was tested in HEK293 cells (n = 3) by immunoprecipitations with anti-DYRK1A followed by an immunoblot using anti-phospho-HIPK2 as described in Widowati et al. [33]. DYRK1A phospho-Tyr321 levels were normalized with DYRK1A total level (orange dashes). Variant DYRK1A phospho-Tyr321 levels were normalized with total DYRK1A protein levels and expressed as percentage of wild-type level. One-way ANOVA was performed to compare variants to wild-type DYRK1A levels. ns not significant; ***p < 0.001; error bars represent SEM. (c) Immunofluorescence experiment showing that Ser660fs (alias Ser660Profs*43) variant leads to DYRK1A protein aggregation when overexpressed in HeLa cells, using a FLAG-tagged DYRK1A proteins carrying Ser660Profs*43. No aggregation was observed for the Ser660* variant.
Identification of a DNAm signature associated with DYRK1A pathogenic variants
To determine if DYRK1A is associated with specific changes in genome-wide DNA methylation (DNAm) in blood, we used methylation array analysis. We compared DNAm in blood for a subset (i.e., the discovery subset) of our cohort carrying pathogenic LoF variants in DYRK1A with age- and sex-matched neurotypical controls and identified n = 402 differentially methylated CpG sites corresponding to 165 RefSeq genes (Table S4, Fig. 3a–b). The sensibility and specificity of the score (0–1) derived from this signature was validated using additional individuals with DYRK1A truncating variants (validation subset), additional controls, and individuals with pathogenic variants in other genes (Table S5; Fig. 3c). Next, we scored the samples with missense variants and found that six classified positively (p.Asp287Val, p.Ser311Phe, p.Arg467Gln, p.Gly168Asp, p.Ile305Arg, and p.Ser324Arg) and three negatively (p.Arg255Gln, p.Tyr462His, p.Thr588Asn) (Fig. 3b–c, Table S5, Figure S9). The sample with the distal frameshift variant p.Ser660fs classified as DNAm positive with a high score (0.92). The sample with the p.Gly486Asp variant clustered out from both DYRK1A cases and controls and its methylation profile was even opposite to DYRK1A LoF cases (increased methylation at sites decreased in LoF cases and vice versa, Figure S9, S11), suggesting this variant might have a gain-of-function (GoF) effect. A notable feature of these GoF CpG sites is that they tended to cluster together, as for instance in the HIST1H3E promoter (Table S4).

(a) Heatmap showing the hierarchical clustering of discovery DYRK1A LoF cases (n = 10) and age- and sex-matched neurotypical discovery controls (n = 24) used to identify the 402 differentially methylated signature sites shown. Each row corresponds to a CpG site differentially methylated (DM) and the color gradient represents the normalized DNA methylation (DNAm) value from -2.0 (blue) to 2.0 (yellow) at each site. DNA methylation at these sites clearly separate discovery cases (gray) from discovery controls (blue). Euclidean distance metric is used for the clustering dendrogram. (b) Principal components analysis (PCA) visualizing the DNAm profiles of the study cohort at the 402 signature sites. Validation of DYRK1A LoF cases (not used to define the signature sites; red) cluster with discovery cases, while missense (yellow) and distal LoF (green) variants cluster with either cases or controls. individual 33 (Gly486Asp) has an opposite DNAm profile to DYRK1A LoF cases at these sites, suggesting a GoF. (c) Support vector machine (SVM) classification model based on the DNA methylation values in the discovery groups. Each sample is plotted based on its scoring by the model. All samples are clearly positive (>0.5) or negative (<0.5). All DYRK1A validation cases from our cohort (n = 6) classified positively, all control validation cases (n = 94) classified negatively. Missense variants classified clearly positively or negatively (yellow), the distal frameshift variant (individual 18, c.1978del) (green), analyzed in duplicate, classified positively. Pathogenic ARID1B (Coffin–Siris syndrome) and KMT2A (Wiedemann–Steiner syndrome) also classified negatively.
Integration of the different tools to reclassify variants
We integrated the clinical score, in silico predictions, functional assays results, and DNAm score to evaluate the pathogenicity of the variants and reclassify them according to ACMG/AMP categories (Fig. 4, Table S6). We found that variants p.Gly168Asp, p.Asp287Val, p.Ile305Arg, p.Ser311Phe, and p.Arg467Gln, identified in individuals with intermediate to high CSDYRK1A scores, led to reduced protein level as well as an absence of autophosphorylation activity, which was previously described for three of them [33, 37]. All classified as DNAm positive, definitively supporting their pathogenicity. For the p.Ser324Arg variant, identified de novo in a patient with an intermediate CSDYRK1A score, we observed only a slight decrease of DYRK1A stability and a partial decrease of its autophosphorylation ability. The binary nature of the DNAm signature, showing a positive score, definitively supports its pathogenic effect.

Representation of the DYRK1A protein (the kinase domain is indicated in red and the catalytic domain in dark red) with the positions of the different variants tested with the sample ID of the individuals indicated inside the circles. Number: number of individuals with ID reported with the variant; gAD: variant reported in individuals from gnomAD; CSDYRK1A poorly (white), intermediate (gray) or highly (black) evocative, or unknown (-); white star indicates that the individual presents a high CSDYRK1A score but also additional clinical manifestations unusual for DYRK1A syndrome; CADD below 25 (white), between 25 and 30 (gray) or above 30 (black); conservation: highly conserved V = 100%, M > 90%, O > 80% (black), moderately V = 100%, M > 90%, O < 80% (gray) or mildly V = 100% M < 90%, O < 80% (white); Expression or autophosphorylation being normal (white), intermediate decreased (gray), strongly decreased (black); localization was normal (white), affected (gray), or not tested (-); DNA methylation positive (black), negative (white), suggestive of a gain-of-function (GoF) effect (hashed) or not tested (-). Final classification: pathogenic (P), benign/likely benign (B), unknown significance (U).
The p.Arg255Gln and p.Tyr462His variants were identified in individuals with low CSDYRK1A score, who had relatively high CADD score (24 and 29.6) but affected amino acids were not highly conserved. They had no effect on protein level, autophosphorylation, and cellular localization of DYRK1A and were classified DNAm negative, and were therefore both considered as likely benign. While parental DNA was not available to test the inheritance of p.Arg255Gln, the variant p.Tyr462His occurred de novo. This individual has an affected brother who does not carry the variant, and exome sequencing of the whole family failed to identify additional promising variants, even taking into account the possibility of two different origins for the brothers. This remains puzzling and thus genome sequencing is ongoing for both. Another de novo variant affecting the same position (p.Tyr462Cys) was identified in a girl with mild developmental delay, hypotonia, and hypermobility without facial dysmorphia, who finally obtained another molecular diagnosis (personal communication, Sander Stegmann, Maastricht University Medical Center). The p.Thr588Asn variant, previously reported as likely pathogenic [6], affects a mildly conserved amino acid and appears to have no effect on mRNA or protein level and function, as described by others [33, 37]. To go further, we tested the ability of the mutant Thr588Asn DYRK1A to phosphorylate MAPT on its Thr212 and confirmed it does not affect its kinase activity (Figure S10). Moreover, a knock-in mouse model generated for Thr588Asn failed to present any decrease of kinase activity and any obvious behavioral phenotype (Supplementary Text, Figure S12). In addition, if this variant was not reported in gnomAD, two other amino acid changes are reported at the same position: p.Thr588Pro (44 times including once at the homozygote state) and p.Thr588Ala (once). All these arguments plus the fact that the patient’s DNAm signature was negative were convincing enough to reclassify this variant as likely benign. The fact that it occurred de novo in a girl with a high CSDYRK1A (15.5/20) remains puzzling, while no additional promising variants were identified in trio exome sequencing data and no positive classification was found using ~20 DNAm signatures available. However, the girl also presents additional manifestations unusual for DYRK1A syndrome such as truncal obesity.
Only one sample showed a DNAm profile different from both controls and individuals with DYRK1A syndrome (Fig. 3a, Figure S9 and S11). This individual has a low CSDYRK1A, presenting relative macrocephaly and ASD without ID and carries a de novo p.Gly486Asp variant. This variant was previously reported in another individual with NDD [38], but it was not possible to obtain DNA, clinical, or inheritance information. No significant change in protein and autophosphorylation level was observed for this variant and analysis of MAPT Thr212 phosphorylation failed to confirm the potential GoF effect (Figure S10).
We characterized the consequences of a distal frameshift de novo variant, p.Ser660fs. Its overexpression leads to cytoplasmic aggregation of DYRK1A, which makes it difficult to quantify the real effect on protein level, autophosphorylation, or kinase activity (Figure S8 and S10). However, its DNAm overlaps those of other individuals with truncating variants located further upstream in the protein, confirming its pathogenic effect (Figure S9). To test if these aggregations could be driven by the novel C-terminal extension added by the frameshift variant (43 amino acids), we introduced nonsense variants at the same position (Ser660* and Ser661*). As no aggregate was detected (Fig. 2c and S8D), we concluded that the C-terminal extension is responsible for the self-aggregation of the mutant DYRK1A protein. Interestingly, the two truncating variants Ser660* and Ser661* did not affect DYRK1A level, localization, or autophosphorylation (Figure S8A, B).
DISCUSSION
Here we report clinical manifestations of 34 novel patients with clear LoF variants in DYRK1A, refining the clinical spectrum associated with DYRK1A syndrome. We used recurrent signs present in individuals to establish a clinical score, which may seem outdated in the era of pangenomic approaches but is in fact very useful to interpret VUS identified by these approaches. Indeed, here we demonstrated that the combination of clinical data together with in silico and in vitro observations are essential to interpret variants accurately.
Since DYRK1A is a highly conserved gene in vertebrates, we assumed that in silico predictive tools using conservation calculated mainly from vertebrates might overestimate the potential pathogenicity of missense variants. We showed that deeper conservation analyses using additional taxa are useful to improve the predictions for missense variants. However, in silico analyses have their limitations, and functional assays are essential to assess variant effect conclusively. We therefore tested the effect of 17 variants and showed that ten of them decreased both DYRK1A protein level and DYRK1A autophosphorylation level. The remaining variants showed no effect on protein function (Fig. 4). However, the absence of effects observed during series of functional tests does not totally exclude a potential effect.
Over the past five years, several studies have found patients with specific monogenic disorders involving genes encoding epigenetic regulatory proteins are associated with DNAm signatures in blood. The advantage of such signatures is the high rate of clear classification (positive/pathogenic vs. negative/benign) they provide for variants. Considering the potential role played by DYRK1A in epigenetic regulation [22, 23, 39], we tested whether DYRK1A LoF leads to such a DNAm profile and identified a DYRK1A DNAm signature with high sensitivity and specificity (Fig. 3). We undertook this work in whole blood (as most signature work is done) due to its clinically availability and in silico tools to account for cell proportion differences. We expect many of these changes to be blood-specific in patients with DYRK1A pathogenic variants. However, enough DNAm changes may overlap other tissues for the blood signature to have cross-tissue utility for variant classification, as we found for fibroblasts in Sotos syndrome [18]. The combination of clinical score (CSDYRK1A), in silico predictions, functional assays, and DNAm signature allow to reclassify ten missense variants as pathogenic. Three variants were considered as likely benign: a variant located in the catalytic domain whose inheritance was unknown, p.Arg255Gln, and two de novo variants located at the end or outside of this domain: p.Tyr462His and p.Thr588Asn.
Still based on methylation data, we suspected a GoF effect for another de novo variant located outside the catalytic domain: p.Gly486Asp. We have already shown that DNAm profiles at gene-specific signature sites provide a functional readout of variant’s effect including GoF activity. Indeed, in previous work, we found the same pattern for a patient with a missense variant in EZH2, typically associated with Weaver syndrome. The patient, presenting undergrowth rather than the overgrowth characteristic of Weaver syndrome, had an opposite DNAm profile to EZH2 cases relative to controls and carried a missense variant that was demonstrated to increase EZH2 activity [19]. In our case, we could not confirm the putative GoF effect of Gly486Asp by measuring MAPT Thr212 phosphorylation. Arranz et al. observed, on the contrary, an increase of DYRK1A kinase activity [37] for this variant, but they reported significant increase for five additional variants, including one present four times in gnomAD (Arg528Trp), which might question the sensitivity of the test.
We identified a de novo distal frameshift variant in the last exon of DYRK1A leading to DYRK1A aggregation in vitro, which needs to be confirmed in vivo. Interestingly, nonsense changes introduced at this position (aa660 and 661) lead to the expression of a protein that seems to be stable, does not aggregate, and maintains its autophosphorylation capacity and ability to phosphorylate MAPT (Fig. 2c, S8C, D, S10). Therefore, we think that distal truncating variants should be interpreted with caution, especially when they escape to NMD. Three additional such distal variants are reported in individuals with ID/NDD in ClinVar and literature (Table S8) for which DNAm analysis on blood samples would be interesting to perform.
In conclusion, we developed a combination of tools efficient to interpret variants identified in DYRK1A. We showed that missense variants located outside and inside the catalytic domain as well as variants leading to distal premature stop codon are not necessarily pathogenic. These results illustrate that variants in DYRK1A, as well as in other NDD causative genes, should be interpreted with caution, even if they occur de novo. In the future, we recommend performing DNAm analysis if blood DNA sample is available or, if not, in vitro testing of variant effect on DYRK1A autophosphorylation.
Data availability
Variants were submitted to ClinVar or to DECIPHER (as indicated in Table 1). Additional data are available upon request.
References
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8. https://doi.org/10.1038/nature21062
Gonzalez-Mantilla AJ, Moreno-De-Luca A, Ledbetter DH, Martin CL. A cross-disorder method to identify novel candidate genes for developmental brain disorders. JAMA Psychiatry. 2016;73:275–83. https://doi.org/10.1001/jamapsychiatry.2015.2692
Møller RS, Kübart S, Hoeltzenbein M, Heye B, Vogel I, Hansen CP, et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am J Hum Genet. 2008;82:1165–70. https://doi.org/10.1016/j.ajhg.2008.03.001
Courcet J-B, Faivre L, Malzac P, Masurel-Paulet A, Lopez E, Callier P, et al. The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J Med Genet. 2012;49:731–36.
van Bon BW, Coe BP, Bernier R, Green C, Gerdts J, Witherspoon K, et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol Psychiatry. 2016;21:126–132. https://doi.org/10.1038/mp.2015.5
Bronicki LM, Redin C, Drunat S, Piton A, Lyons M, Passemard S, et al. Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. Eur J Hum Genet. 2015;23:1482–1487. https://doi.org/10.1038/ejhg.2015.29
Ruaud L, Mignot C, Guët A, Ohl C, Nava C, Héron D, et al. DYRK1A mutations in two unrelated patients. Eur J Med Genet. 2015;58:168–174. https://doi.org/10.1016/j.ejmg.2014.12.014
Ji J, Lee H, Argiropoulos B, Dorrani N, Mann J, Martinez-Agosto JA, et al. DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur J Hum Genet. 2015;23:1473–1481. https://doi.org/10.1038/ejhg.2015.71
Meissner LE, Macnamara EF, D’souza P, Yang J, Vezina G, Undiagnosed Diseases N. et al. DYRK1A pathogenic variants in two patients with syndromic intellectual disability and a review of the literature. Mol Genet Genomic Med. 2020;7:e1544. https://doi.org/10.1002/mgg3.1544
O’roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. https://doi.org/10.1126/science.1227764
Earl RK, Turner TN, Mefford HC, Hudac CM, Gerdts J, Eichler EE, et al. Clinical phenotype of ASD-associated DYRK1A haploinsufficiency. Mol Autism. 2017;8:54. https://doi.org/10.1186/s13229-017-0173-5
Himpel S, Panzer P, Eirmbter K, Czajkowska H, Sayed M, Packman LC, et al. Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem J. 2001;359:497–505. https://doi.org/10.1042/0264-6021:3590497
Hämmerle B, Elizalde C, Tejedor FJ. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. Eur J Neurosci. 2008;27:1061–1074. https://doi.org/10.1111/j.1460-9568.2008.06092.x
Tejedor FJ, Hämmerle B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2011;278:223–235. https://doi.org/10.1111/j.1742-4658.2010.07954.x
Duchon A, Herault Y. DYRK1A, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in Down syndrome. Front Behav Neurosci. 2016;10:104. https://doi.org/10.3389/fnbeh.2016.00104
Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Wang X, et al. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J. 2001;355:609–615. https://doi.org/10.1042/bj3550609
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. https://doi.org/10.1038/gim.2015.30
Choufani S, Cytrynbaum C, Chung BH, Turinsky AL, Grafodatskaya D, Chen YA, et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat Commun. 2015;6:10207. https://doi.org/10.1038/ncomms10207
Choufani S, Gibson WT, Turinsky AL, Chung B, Wang T, Garg K, et al. DNA methylation signature for EZH2 functionally classifies sequence variants in three PRC2 complex genes. Am J Hum Genet. 2020;106:596–610. https://doi.org/10.1016/j.ajhg.2020.03.008
Chater-Diehl E, Ejaz R, Cytrynbaum C, Siu MT, Turinsky A, Choufani S, et al. New insights into DNA methylation signatures: SMARCA2 variants in Nicolaides-Baraitser syndrome. BMC Med Genomics. 2019;12:105. https://doi.org/10.1186/s12920-019-0555-y
Aref-Eshghi E, Bend EG, Colaiacovo S, Caudle M, Chakrabarti R, Napier M, et al. Diagnostic utility of genome-wide DNA methylation testing in genetically unsolved individuals with suspected hereditary conditions. Am J Hum Genet. 2019;104:685–700. https://doi.org/10.1016/j.ajhg.2019.03.008
Jang SM, Azebi S, Soubigou G, Muchardt C. DYRK1A phoshorylates histone H3 to differentially regulate the binding of HP1 isoforms and antagonize HP1-mediated transcriptional repression. EMBO Rep. 2014;15:686–694. https://doi.org/10.15252/embr.201338356
Li S, Xu C, Fu Y, Lei PJ, Yao Y, Yang W, et al. DYRK1A interacts with histone acetyl transferase p300 and CBP and localizes to enhancers. Nucleic Acids Res. 2018;46:11202–11213. https://doi.org/10.1093/nar/gky754
Redin C, Gérard B, Lauer J, Herenger Y, Muller J, Quartier A, et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet. 2014;51:724–736. https://doi.org/10.1136/jmedgenet-2014-102554
Carion N, Briand A, Cuisset L, Pacot L, Afenjar A, Bienvenu T. Loss of the KH1 domain of FMR1 in humans due to a synonymous variant causes global developmental retardation. Gene. 2020;753:144793. https://doi.org/10.1016/j.gene.2020.144793
Nasser H, Vera L, Elmaleh-Bergès M, Steindl K, Letard P, Teissier N, et al. CDK5RAP2 primary microcephaly is associated with hypothalamic, retinal and cochlear developmental defects. J Med Genet. 2020;57:389–399. https://doi.org/10.1136/jmedgenet-2019-106474
Balak C, Benard M, Schaefer E, Iqbal S, Ramsey K, Ernoult-Lange M, et al. Rare de novo missense variants in RNA helicase DDX6 cause intellectual disability and dysmorphic features and lead to P-body defects and RNA dysregulation. Am J Hum Genet. 2019;105:509–525. https://doi.org/10.1016/j.ajhg.2019.07.010
Quartier A, Chatrousse L, Redin C, Keime C, Haumesser N, Maglott-Roth A, et al. Genes and pathways regulated by androgens in human neural cells, potential candidates for the male excess in autism spectrum disorder. Biol Psychiatry. 2018;84:239–252. https://doi.org/10.1016/j.biopsych.2018.01.002
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. https://doi.org/10.1093/bioinformatics/btp033
Kress A, Lecompte O, Poch O, Thompson JD. PROBE: analysis and visualization of protein block-level evolution. Bioinformatics. 2018;34:3390–3392. https://doi.org/10.1093/bioinformatics/bty367
Mattioli F, Isidor B, Abdul-Rahman O, Gunter A, Huang L, Kumar R, et al. Clinical and functional characterization of recurrent missense variants implicated in THOC6-related intellectual disability. Hum Mol Genet. 2019;28:952–960. https://doi.org/10.1093/hmg/ddy391
Quartier A, Courraud J, Thi Ha T, McGillivray G, Isidor B, Rose K, et al. Novel mutations in NLGN3 causing autism spectrum disorder and cognitive impairment. Hum Mutat. 2019;40:2021–2032. https://doi.org/10.1002/humu.23836
Widowati EW, Ernst S, Hausmann R, Müller-Newen G, Becker W Functional characterization of DYRK1A missense variants associated with a syndromic form of intellectual deficiency and autism. Biol Open. 2018;7. https://doi.org/10.1242/bio.032862
Lee K-S, Choi M, Kwon D-W, Kim D, Choi JM, Kim AK, et al. A novel de novo heterozygous DYRK1A mutation causes complete loss of DYRK1A function and developmental delay. Sci Rep. 2020;10:9849. https://doi.org/10.1038/s41598-020-66750-y
Blackburn ATM, Bekheirnia N, Uma VC, Corkins ME, Xu Y, Rosenfeld JA, et al. DYRK1A-related intellectual disability: a syndrome associated with congenital anomalies of the kidney and urinary tract. Genet Med. 2019;21:2755–2764. https://doi.org/10.1038/s41436-019-0576-0
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. https://doi.org/10.1038/ng.2892
Arranz J, Balducci E, Arató K, Sánchez-Elexpuru G, Najas S, Parras A, et al. Impaired development of neocortical circuits contributes to the neurological alterations in DYRK1A haploinsufficiency syndrome. Neurobiol Dis. 2019;127:210–222. https://doi.org/10.1016/j.nbd.2019.02.022
Dang T, Duan WY, Yu B, Tong DL, Cheng C, Zhang YF, et al. Autism-associated Dyrk1a truncation mutants impair neuronal dendritic and spine growth and interfere with postnatal cortical development. Mol Psychiatry. 2018;23:747–758. https://doi.org/10.1038/mp.2016.253
Lepagnol-Bestel A-M, Zvara A, Maussion G, Quignon F, Ngimbous B, Ramoz N, et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum Mol Genet. 2009;18:1405–1414. https://doi.org/10.1093/hmg/ddp047
Acknowledgements
The authors thank the families for their participation and support. The authors also thank the Agence de Biomédecine, Fondation APLM, Fondation Maladies Rares and Fondation Jérome Lejeune for financial support. We also thank the Centre National de Génotypage, the diagnostic laboratories of Hôpitaux Universitaire de Strasbourg (HUS), clinical genetics residents, the GenomEast sequencing and the molecular biology platforms of IGBMC.
WEB RESOURCES
The URLs for online tools and data presented herein are:
CADD: https://cadd.gs.washington.edu/
ClinVar: http://www.ncbi.nlm.nih.gov/ClinVar/
Clustal Omega: https://www.ebi.ac.uk/Tools/msa/clustalo/
dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP/
Decipher: https://decipher.sanger.ac.uk/
gnomAD : http://gnomAD.broadinstitute.org/
Integrative Genomics Viewer (IGV): http://www.broadinstitute.org/igv/
Mutation Nomenclature: http://www.hgvs.org/mutnomen/recs.html
Nnsplice: https://www.fruitfly.org/seq_tools/splice.html
OMIM: http://www.omim/org/
UCSC: http://genome.ucsc.edu/
OrthoInspector database: https://www.lbgi.fr/orthoinspectorv3/databases
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declaration
Individuals were referred by clinical geneticists for genetic testing as part of routine clinical care. All patients enrolled and/or their legal representative signed informed consent for research use and authorization for publication, including photograph publication for those included in Figure S3. All the institutions received local institutional review board (IRB) approval to use these data in research. The main IRB approval was obtained from the Ethics Committee of the Strasbourg University Hospital (CCPPRB). Experiments performed on animals were all done in compliance with the ARRIVE Guidelines and were noninvasive procedures approved by Com’Eth, the local ethics committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Courraud, J., Chater-Diehl, E., Durand, B. et al. Integrative approach to interpret DYRK1A variants, leading to a frequent neurodevelopmental disorder. Genet Med 23, 2150–2159 (2021). https://doi.org/10.1038/s41436-021-01263-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01263-1
This article is cited by
-
Molecular consequences of PQBP1 deficiency, involved in the X-linked Renpenning syndrome
Molecular Psychiatry (2023)
-
Social motivation a relative strength in DYRK1A syndrome on a background of significant speech and language impairments
European Journal of Human Genetics (2022)
-
Differential maturation and chaperone dependence of the paralogous protein kinases DYRK1A and DYRK1B
Scientific Reports (2022)
