
Abstract
Carrier screening began 50 years ago with screening for conditions that have a high prevalence in defined racial/ethnic groups (e.g., Tay–Sachs disease in the Ashkenazi Jewish population; sickle cell disease in Black individuals). Cystic fibrosis was the first medical condition for which panethnic screening was recommended, followed by spinal muscular atrophy. Next-generation sequencing allows low cost and high throughput identification of sequence variants across many genes simultaneously. Since the phrase “expanded carrier screening” is nonspecific, there is a need to define carrier screening processes in a way that will allow equitable opportunity for patients to learn their reproductive risks using next-generation sequencing technology. An improved understanding of this risk allows patients to make informed reproductive decisions. Reproductive decision making is the established metric for clinical utility of population-based carrier screening. Furthermore, standardization of the screening approach will facilitate testing consistency. This practice resource reviews the current status of carrier screening, provides answers to some of the emerging questions, and recommends a consistent and equitable approach for offering carrier screening to all individuals during pregnancy or preconception.
Similar content being viewed by others

INTRODUCTION
Carrier screening is used to identify individuals or couples that are at risk to have a child with an autosomal recessive or X-linked genetic disorder. Throughout this document, the term “carrier” specifically refers to individuals who are heterozygous for a pathogenic or likely pathogenic variant in an autosomal recessive or X-linked condition. Once identified, carriers of these disorders can become educated about their risks and consider a range of reproductive options. Historically, criteria for screening have included: phenotype severity that may impact decision making,1,2 high prevalence of carriers in the screened population,2 established analytic validity of screening methods,2,3 predictable genotype–phenotype correlation,2 available prenatal diagnosis and reproductive options.2,4 Although general principles remain similar today these do not speak to the genes that should be included as part of routine carrier screening.
In 2013, the American College of Medical Genetics and Genomics (ACMG) linked the utility of carrier screening to reproductive decision making.1 Decision making is inherently tied to the severity of any condition being screened. This consensus group recognized that there will be disagreement when defining the severity of various conditions. However, we used published definitions which include (1) profound: shortened lifespan during infancy or childhood, intellectual disability; (2) severe: death in early adulthood, impaired mobility or a [disabling] malformation involving an internal organ; (3) moderate: neurosensory impairment, immune deficiency or cancer, mental illness, dysmorphic features; and (4) mild: not meeting one of those described.5
Carrier screening for heritable autosomal recessive conditions, which began 50 years ago,6 targeted at-risk populations who have been traditionally defined as an ethnic group that is geographically isolated or one with cultural norms and customs that limit random mating (Ashkenazi Jewish [AJ], Amish, Hutterites). The successful implementation of biochemical screening for Tay–Sachs disease (TSD) among the AJ population in the 1970s7 paved the way to consider carrier screening for other disorders. TSD, a condition meeting the definition for profound severity, has a carrier frequency of approximately 1/30 among AJ and 1/300 among the general population.8 Similarly, sickle cell disease has a long history of screening.9 It has a carrier frequency of approximately 1/13 among “African-American[s]” and 1/20 in “Hispanic[s]” resulting in a carrier frequency of about 1/66 in the general population.10 A wide range in the carrier frequencies of genetic conditions between at-risk groups and the general population raises questions of equity when implementing carrier screening. It raises concerns over how screening policies impact information that leads to reproductive decision making. Restricting carrier screening by using socially defined ethnic constructs or by self-identified ancestry is both inequitable and scientifically flawed. Importantly, those who self-identify with a specific race/ethnicity may be at odds with ancestry defined genetically, which is of relevance to carrier screening.11,12 A recent report demonstrated that relying on self-identification of AJ ancestry as a criteria to screen for conditions common in the AJ population is imperfect.13 It is important that carrier screening goes beyond commonly recognized at-risk groups and includes diverse populations.
The goals of carrier screening have not changed over time. However, the technology used in carrier screening has changed dramatically allowing for high throughput with rapid turnaround times.14 As the cost of sequencing the entire genome has fallen,15,16 so too have the costs of sequencing panels of genes. The American College of Medical Genetics and Genomics (ACMG)’s last official documents regarding carrier screening for specific conditions were published in 2004 and 2008.17,18 ACMG adopted an ethnic and population neutral approach to carrier screening for cystic fibrosis and spinal muscular atrophy.17,18 The American College of Obstetricians and Gynecologists (ACOG) also endorsed universal screening for these two conditions and suggested that one additional screening criterion might be a carrier frequency of ≥1/100.19 Recommendations by ACMG predate advances in gene sequencing technology. Moreover, there is now a greater societal awareness over equity in care that has evolved since ACOG and ACMG published statements on carrier screening.20 Whereas in prior years, carrier screening was a scarce resource reserved only for those with the highest risk; a more attainable price point now allows for the opportunity to reach every patient.
In 2015, the ACMG, along with other professional organizations, published a Points to Consider joint statement focused on expanded carrier screening21 wherein general genetic principles and a historical perspective were discussed. An emphasis was placed on the consent process including elements of pre- and post-test counseling. The principles emphasized in that document remain important today. This current document considers more recent published information and closes gaps in the previously published Points to Consider while acknowledging technological advances in sequencing and the need for equity and distributive justice of genomic technologies. This document replaces the ACMG position statement on prenatal/preconception expanded carrier screening.1
METHODS
This consensus group convened to develop and answer a series of questions that are important for clinicians and reproductive age patients to consider as part of the carrier screening process (Box 1).
RESULTS AND DISCUSSION
Consensus question 1: Are analytical and clinical validity established for carrier screening?
Analytical validity refers to how well the test predicts the presence or absence of a particular genetic change, which encompasses sensitivity, specificity, and accuracy among other factors.22 Carrier screening relies on laboratory methods such as next-generation sequencing (NGS), polymerase chain reaction (PCR), Sanger sequencing, multiplex ligation-dependent probe amplification (MLPA), microarray, and other methods to identify small-scale genetic changes including single-nucleotide variants (SNVs), and large-scale structural variants (SVs), including copy-number variants (CNVs). It is important that laboratories put in place effective quality metrics within the various testing platforms used, to ensure accuracy of variants detected to prevent false negative and/or false positive calls. The ACMG has established guidelines for the development of NGS assays.23 Each test method optimized for clinical use, should undergo robust validation processes as required by the Clinical Laboratory Improvement Amendments (CLIA) and the College of American Pathologists (CAP) to define the analytical sensitivity, analytical specificity, and analytical accuracy of an assay that establishes confidence in the detection, analysis, and reporting of genetic variants. Analytical validity is in part a function of the number of variants and number of genes interrogated. Interrogations of greater numbers of either variants or variants and genes has the potential for greater error; however, the CLIA validation process mitigates this concern.
Clinical validity relates a test’s result to the condition for which the test is designed addressing the issue of how well the genetic variant being analyzed is related to the presence, absence, or risk of a specific disease.22 In other words, a test has robust clinical validity when both the negative and positive predictive values are high.24,25 Genetic variants are classified as either pathogenic, likely pathogenic, uncertain significance, likely benign, or benign.26 Generally, in the setting of screening, laboratories report only those variants that are classified as pathogenic (>99% certainty) or likely (>90% certainty) pathogenic. However, there are exceptions leading to instances where a variant of uncertain significance (VUS) is reported.27 For example, when one member of a couple is known to carry a pathogenic or likely pathogenic variant, reporting a VUS after screening the second member of the couple may be considered.27 The preconception counseling session ideally addresses return of results when a VUS is identified. It is important for patients to understand that changes in the interpretation of clinical genomic test results are possible and recontact may be important. Furthermore, when medical or family history changes this should be communicated with the patient’s care provider.28
Carrier screening cannot completely eliminate the risk of being a carrier of a heritable condition, because:
-
All genes that cause a condition may not be known.
-
All genes that cause a condition may not be examined.
-
Causative variants may be in a region not included in the test.
-
Causative variants may be undetectable by the technology/analysis employed.
-
Analysis of gene sequence and its structural variants may be technically difficult.
-
Variants may be misclassified with regard to pathogenicity (e.g., laboratory’s algorithm for classification of variants).
An individual’s residual risk to be a carrier after having a negative screening test can be calculated as follows: Population Carrier frequency × (1 – Detection Rate). However, when carrier screening is implemented by simultaneously interrogating multiple variants within multiple genes for rare conditions, the carrier frequency and detection rate may not be known for each condition being screened. It is impractical to provide a precise residual risk after carrier screening that includes simultaneous analysis of multiple uncommon or rare variants within genes. Instead, patients should be aware that a negative screening test does not eliminate the risk of being a carrier for any condition (i.e., gene variant), although this risk is greatly reduced.
Carrier screening aims to identify pathogenic and likely pathogenic variants within genes known to cause a condition or phenotype of interest as underscored by the relationship between ClinVar and ClinGen. ClinVar29 is a national registry for the classification of variants within genes. All laboratories that perform genetic testing are expected to report variants identified within their testing cohort using specific submission guidelines to ensure consistency. ClinGen30,31 hosts a gene-level database (https://www.clinicalgenome.org) that displays results from its gene curation expert panels which score the association of a gene with a condition or phenotype. One of seven classes are used to describe this association: no evidence reported, refuted, disputed, limited, moderate, strong, definitive. Documenting case observations to support these associations relies on clinical information obtained through medical history, pedigree analysis, laboratory data, pathology studies, imaging, and physical examination.25 It is easy to understand why conditions characterized by variable expressivity or reduced penetrance may produce a lower gene–disease association score. Either of these may make the clinical tools used to define a condition unreliable. For example, reduced penetrance may limit the value of pedigree analysis. Variable expressivity may cause difficulty in linking a physical exam finding to a genetic diagnosis. Sometimes a gene is associated with more than one condition, so within ClinGen a gene may be classified according to more than one clinical condition.
In summary:
-
Analytical validity of carrier screening is to be established by a laboratory in compliance with CLIA/CAP regulations and adhering to ACMG Laboratory Standards and Guidelines.
-
Establishing clinical validity is gene and condition specific. For example, CFTR and many (but not all) of its variants are associated with cystic fibrosis.27
-
As evidence evolves, ClinVar and ClinGen continually update pathogenicity of variants and the association between genes and conditions, respectively.
-
A negative screening result does not eliminate the risk of being a carrier for the conditions screened but does reduce that risk. The residual risk to be a carrier for any condition is never zero.
-
It is not practical to generate a precise residual risk estimate for the group of conditions interrogated through multiplex screening after a negative screening result. This requires a defined carrier frequency and detection rate for all conditions screened.
Consensus question 2: Has clinical utility been established for carrier screening?
Clinical utility in its narrowest sense refers to the ability of a screening or diagnostic test to prevent or ameliorate adverse health outcomes such as mortality, morbidity, or disability through the adoption of efficacious treatments conditioned on test results.32 The considerations that determine clinical utility are (1) whether the test and any subsequent interventions lead to an improved health outcome among people with a positive test result; and (2) what risks occur as a result of testing.25 Importantly, the specific metric used to measure clinical utility is context specific. For carrier screening, clinical utility is measured by the fact that individuals or couples are informed and may alter reproductive decision making because of the carrier screening results.33,34,35
The clinical utility of carrier screening is represented by its ability to provide individuals an opportunity to discuss their risks and consider reproductive options that are available prepregnancy, during pregnancy, or after birth. Availability of reproductive options may depend on various socioeconomical, legal, and cultural factors in different regions. Examples of reproductive options include:
-
In vitro fertilization with preimplantation genetic testing for monogenic conditions.
-
Use of donor gamete/embryo.
-
Adoption.
-
Prenatal diagnosis using chorionic villus sampling or amniocentesis followed by a decision to either prepare for an affected child including special care after birth or terminate the pregnancy.
-
A decision not to have children.
Studies have established that carrier screening of many conditions simultaneously does have an impact on reproductive decision making. Although these studies are few and represent survey data, they include more than 470,000 screened patients.25,34,35,36,37 In the two largest studies (April 2014 through August 2015 and September 2015 through 2017), there were 110 and 176 genes analyzed, respectively. The response rates varied, but of those responding, a majority (~60%) took some action in response to being identified as an at-risk couple. In these studies, reproductive decision making was more common when patients received results before an established pregnancy (62–77%). The most common decisions in the largest study were to pursue in vitro fertilization with preimplantation genetic diagnosis (59%), undergo a diagnostic test during pregnancy (20%), and use of a donor gamete (7.7%). Adoption was being considered by 5.1% at the time survey data were collected.35 In the two largest studies, an affected fetus was identified in 16% (3/19) to 36% (20/56) of those having a diagnostic procedure and 67% (2/3) and 40% (8/20) respectively discontinued their pregnancy.34,35 This workgroup acknowledges that studies listed above may not reflect the clinical utility in an ethnically diverse population of individuals seeking carrier testing. We encourage additional ethnically inclusive studies to address this issue in the future.
In summary:
-
Carrier screening enables those screened to consider their reproductive risks, reproductive options, and to make informed decisions.
-
Published evidence supports clinical utility for carrier screening of multiple conditions simultaneously.33
Consensus question 3: Is “expanded carrier screening” a precise term?
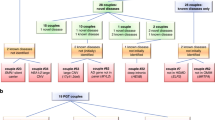
Expanded carrier screening is not well or precisely defined by professional organizations.1,2,19,21 The term “expanded” might imply an increased number of genes, or a paradigm shift from screening populations with higher carrier frequencies to screening those without regard to ancestry, or both. For some, “expanded” may represent screening many more variants within a gene. It is important for patients and health-care professionals to communicate more precisely when speaking about carrier screening by using a precise and consistent language. Some molecular testing laboratories now offer obstetric care professionals “expanded carrier screening” packages that can include more than a thousand genes;38 however, other laboratories screen several hundred and the overlap in genes between laboratories is limited. In practical terms, there is no industry standard when it comes to the number of genes interrogated for carrier screening that is used to inform reproductive decision making. Thus far, molecular testing laboratories have determined the genes/conditions on “expanded” carrier screening panels. We propose adopting a tiered definition of carrier screening model (Fig. 1), which will allow patients and health-care professionals to communicate with greater precision.

*Recommended by the American College of Medical Genetics and Genomics (ACMG)17,18 and American College of Obstetricians and Gynecologists (ACOG).19 ±Recommended by ACOG.2 §Supported by literature.49,50 ¥Offered by molecular testing laboratories; the list of genes/conditions may vary by the laboratory. CF cystic fibrosis, SMA spinal muscular atrophy.
ACMG recommends:
-
The phrase “expanded carrier screening” be replaced by “carrier screening”.
-
Adopting a more precise tiered system based on carrier frequency (Fig. 1).
When patients are asked to report their ancestry, they respond with their learned/self-identified ancestry or report their ethnicity and race. The manner in which patients ascribe their ancestry is impacted by ethnic admixture, awareness and preservation of knowledge about ancestral origins, prevailing ideologies about race and racial divisions, and the number of generations removed from the arrival of immigrant ancestors.39 Ethnic groups are defined by characteristics that include cultural traditions and norms.40 There is increasing evidence that self-described ethnicity has inherent and unpredictable inaccuracies,12,13,41,42,43,44 and genetically determined ancestry using single-nucleotide polymorphisms helps identify population/geographic origin, which is of particular importance for carrier screening. A risk-based strategy of carrier screening, which relied on self-described ethnicity, was first adopted for Tay–Sachs disease screening7 and for the most part continues today.19,21 In many cases reproductive partners are not chosen randomly.45 Instead partners are chosen based on societal pressures, norms, and expectations. However, data show that population intermixing in the United States has increased dramatically over the last several centuries.39 This requires that carrier screening be useful for all of those living in the United States regardless of their ancestry.
ACMG recommends:
-
Carrier screening paradigms should be ethnic and population neutral and more inclusive of diverse populations to promote equity and inclusion.
Consensus question 4: What screening approach should be offered to patients considering carrier screening?
This consensus group recommends establishing a tier-based system of carrier screening, which will enhance communication and precision while advancing equity in carrier screening.
Tier 1 screening conveys the recommendations previously adopted by ACMG17,18 and ACOG.19 Tier 1 screening adopts an ethnic and population neutral approach when screening for cystic fibrosis and spinal muscular atrophy. Beyond these two conditions, additional carrier screening is determined after risk assessment, which incorporates personal medical and family history as well as laboratory and imaging information where appropriate.
Tier 2 carrier screening stems from an ACOG recommendation for conditions that have a severe or moderate phenotype and a carrier frequency of at least 1/100.2 A carrier frequency of at least 1/100 would encompass screening all patients for spinal muscular atrophy (SMA) since SMA carrier frequency was thought to be 1/60 without regard to the population screened.18 Studies have shown that the carrier frequency of SMA in the United States is not uniform across populations. In “Caucasian[s]” (This term is no longer used by the journal but is used in the original article to which these studies refer. We have therefore not changed the term but recognize it does not accurately describe the ancestry of the populations originally studied.46) this has been shown to be 1/46 and in “Hispanic[s]” 1/125.47 For cystic fibrosis when 32 pathogenic variants were examined among a US population, carrier frequency ranged from 1/28 (“Caucasian”) (This term is no longer used by the journal but is used in the original article to which these studies refer. We have therefore not changed the term but recognize it does not accurately describe the ancestry of the populations originally studied.46) to 1/105 (“African American”) and 1/261 (“Asian”).48 These data demonstrate that carrier screening for two common conditions using a carrier frequency threshold of 1/100 may not be equitable across diverse populations. Others have shown that limiting the carrier frequency to ≥1/100 creates missed opportunities to identify couples at risk for serious conditions.49,50
We define Tier 3 screening as carrier screening for conditions with a carrier frequency ≥1/200. The reader is directed to the Supplemental material (“Rationale for Tier 3 Screening” and Figure S1) for a detailed description of the derivation of ≥1/200 as a criterion for autosomal recessive genes. Tier 2 and Tier 3 screening prioritize carrier frequency as a way to think about conditions most appropriate for screening in the general population. However, when ACOG proposed this level, they did not specify whether it was thinking about carrier frequency in terms of the global population or subpopulations. We use “carrier frequency” to mean in any ethnic group with reasonable representation in the United States.
Tier 4 includes genes less common than those in Tier 3 and can identify additional at-risk couples.49,50 Tier 4 has no lower limit carrier screening frequency and can greatly extend the number of conditions screened. Although there are many serious conditions at a carrier frequency below 1/200,49 there may be less information about the natural history of many of these conditions. Additionally, pleiotropy, locus heterogeneity, variant interpretation and poor genotype–phenotype correlation may disproportionately impact the ability to provide accurate prognostic information for these rarer conditions. For these reasons, the clinical validity at this level of carrier screening may be less compelling, therefore we suggest reserving this level of screening for consanguineous pregnancies (second cousins or closer) and in couples where family or medical history suggests Tier 4 screening might be beneficial. Some patients want maximum information and will ultimately choose to have Tier 4 screening either due to convenience (a diagnostic laboratory might make their test the most accessible and hassle-free) or simply because it tests for the most conditions. Importantly, patients should understand that their chance of being a carrier for one or more conditions increases as the number of conditions screened is increased. Also, laboratories may not offer screening for the same genes within the Tier 4 option. Independent of whether laboratories offer conditions that satisfy the carrier frequencies of Tier 2, Tier 3, or Tier 4, all conditions screened should adhere to the same criteria (e.g., at least moderate severity).
ACMG recommends:
-
All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening.
-
Tier 4 screening should be considered:
-
When a pregnancy stems from a known or possible consanguineous relationship (second cousins or closer);
-
When a family or personal medical history warrants.
-
ACMG does not recommend:
-
Offering Tier 1 and/or Tier 2 screening, because these do not provide equitable evaluation of all racial/ethnic groups.
-
Routine offering of Tier 4 panels.
Consensus question 5: Which autosomal recessive conditions are appropriate for carrier screening?
Professional organizations have an obligation to define the conditions appropriate for carrier screening. Until now, molecular testing laboratories have assumed this responsibility with the consequence that conditions screened for are not uniform across laboratories.38 We applied several criteria (Fig. 2) to determine the autosomal recessive genes listed in Tables 1–5.

Criterion for genes listed in Tables 1-4 were identical and derive from gnomAD. Those genes listed in Table 5 do not derive from gnomAD data. The X-linked conditions derive from the OMIM database.55 The prevalence data for X-linked conditions derives from either OMIM,55 Orphanet,63 or MedlinePlus.64 All conditions were at least moderately severe.5,65 OMIM Online Mendelian Inheritance in Man.55.
There were 86 genes that satisfied the aforementioned criteria (Tables 1–4). After reviewing this list of genes, we evaluated genes that previously have been recommended for carrier screening by ACOG or ACMG.44,51 We identified three genes (SMN1: spinal muscular atrophy, ELP1: familial dysautonomia, and BLM: Bloom syndrome) and included these in Table 5. All three of these genes are associated with conditions that have a carrier frequency that is highly represented in one or more patient populations and have the potential to be underrepresented in gnomAD. Detection of SMN1 copy number by NGS is impeded by the presence of a highly homologous pseudogene (SMN2), and could artifactually lower allele frequencies in gnomAD. Like SMN1, the HBA locus is technically complex to assess and most cases of ɑ-thalassemia result from deletions of one or more of the alpha globin genes (HBA1 and HBA2) and thus, could create an artifactually lower allele frequency in gnomAD. The allele frequencies of sequence variants in gnomAD v2.0.2 for ELP1 and BLM were less common than 1/200, but these genes are known to have an allele frequency of at least 1/200 in AJ. Friedreich ataxia is a recessive trinucleotide repeat disorder that is associated with a GAA expansion located in intron 1 of the FXN gene. The condition has its highest carrier frequency in White populations from Northwestern Europe (Spain to Ireland).52 The remaining genes listed in Table 5 have a carrier frequency ≥1/200 in a US subpopulation. Subpopulations included were the AJ and Puerto Rican, each having at least 1% representation in the United States and US territories combined.
In total, we recommend 97 autosomal recessive genes for carrier screening in Tier 3. We cross-referenced Tier 3 autosomal recessive genes to ClinGen30 for gene–disease association. One gene was excluded (BCS1L) because the curation in ClinGen concluded there was “limited” evidence to support a gene–disease association. A commitment to ongoing curation of the autosomal recessive genes will ensure that new information is reflected in the genes recommended for screening in Tier 3 in future iterations. Curation should include technologies available that will ensure high throughput and accurate screening.
Cross-referencing to ClinGen and the ACMG secondary findings list v3.053 allowed for additional observations. Gene–disease association was confirmed as “definitive” in ClinGen for 39 of 97 (40%) (Table S1). Many genes we recommend have not been curated in ClinGen (e.g., CFTR, SMN1, HBB, ARSA). Two genes (MMUT and USH3) we recommend for screening could not be found in ClinGen, likely due to limited curation to date. We also cross-referenced Tier 3 genes to those recommended for universal newborn screening (Table S1). Two genes associated with hearing loss (GJB2 and SLC26A4) are included for screening. We recommend 16 autosomal recessive genes that are screened using metabolic analytes at the time of newborn screening. The potential impact that screening for autosomal recessive conditions will have on families is discussed in the Supplement.
ACMG recommends:
-
All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening for autosomal recessive (Tables 1–5) and X-linked (Table 6) conditions.
Table 6 X-linked genes recommended for carrier screening. -
Reproductive partners of pregnant patients and those planning a pregnancy may be offered Tier 3 carrier screening for autosomal recessive conditions (Tables 1–5) when carrier screening is performed simultaneously with their partner.
-
Ongoing curation of Tier 3 autosomal recessive genes with input from:
-
ACMG Committees and Work Groups;
-
Additional professional organizations and the lay public as appropriate.
-
Consensus question 6: Which X-linked conditions are appropriate for carrier screening?
Some laboratories offer screening for X-linked conditions as part of their carrier screening package. Like autosomal recessive conditions, the X-linked conditions screened do not overlap across the molecular testing laboratories. In fact, some carrier panels on the market contain genes associated with conditions that have a prevalence of 1 in 3,500 while others a condition with a prevalence less than 1 in 1,000,000. It is important that any designated panel include a transparent description of the process used for including/excluding those genes.
The reader is directed to the Supplemental material (“Rationale for Tier 3 screening” and Figure S1) for a detailed description of the derivation of 1/40,000 disease prevalence as a criterion for X-linked gene inclusion. We applied several criteria (Fig. 2) to determine the X-linked conditions listed in Table 6. Based on the aforementioned criteria, we identified 16 genes that are appropriate for carrier screening (Table 6). Cross-referencing these genes to ClinGen revealed that gene–disease association was definitive for 13/16 (81%). The remaining three have not been curated by ClinGen, including DMD, NR0B1, and RPGR. Among X-linked genes, three are on the ACMG secondary findings list v3.0 (ABCD1 [adrenoleukodystrophy], GLA [Fabry disease], and OTC [ornithine transcarbamylase deficiency]).53 The potential impact that screening for X-linked conditions will have on families is discussed in the Supplement. A commitment to ongoing curation of the X-linked genes will ensure that new information is reflected in the genes recommended for screening in Tier 3 in future iterations. Curation should include technologies available that will ensure high throughput and accurate screening.
ACMG recommends:
-
All XX patients should be offered screening for only those X-linked genes listed in Table 6 as part of Tier 3 screening.
-
Ongoing curation of Tier 3 X-linked genes with input from:
-
ACMG Committees and Work Groups;
-
Additional professional organizations and the lay public as appropriate.
-
Consensus question 7: What should the clinician expect with regard to laboratory reporting of carrier screening results?
The clinical laboratory report represents the final postanalytical step of laboratory testing and is a documented communication to the referring clinician. It should be a structured document with clinically significant findings easily identified and understood by the ordering health-care professional. Information should be provided in a clear, concise, and accurate manner that is adherent to regulatory standards (42 CFR § 493.1291). Several ACMG documents address norms and elements of a clinical laboratory report, including report sections, transparency of methods and limitations, standardized five-category variant classifications, and uniform Human Genome Variation Society (HGVS)–based variant annotations.23,26 It is important that the report clearly conveys:
-
ACMG carrier screening tier number and genetic content of the panel with all tested genes and transcripts listed, or, if the number is large, referenced to an accessible website.
-
Whether a targeted (assessment of predefined variants) or comprehensive (assessment of full coding region with splice junctions) approach is carried out with details of the methodology and limitations.
-
Detectable types of DNA variation (e.g., SNVs, CNVs, structural rearrangements).
-
Variant classification range that is used for reporting.
Reporting and interpreting results depends on the clinical context and indication for testing. When results are negative, it is often impractical to provide residual risk estimates because (1) for many of the X-linked genes screened, carrier frequencies are imprecise; (2) data sets and populations used to establish carrier frequency can vary; and (3) calculations depend on the patient’s self-identified ethnicity. However, whenever possible, the analytical sensitivity of detecting different variant types and the detection rate should be provided. This will help to emphasize that a negative test does not eliminate the possibility of being a carrier for any condition screened, but it does reduce this risk.
All pathogenic and likely pathogenic variants should be reported back to the ordering health-care professional. However, a gene-specific comprehensive sequencing approach with the option of reporting of a VUS should be considered for partners of identified carriers27 and discussed during pretest counseling. Reports of positive results should include brief clinical information about the disorder, penetrance if known, and variability in expression if understood. Information about genotype–phenotype correlations may be provided with relevant limitations since correlations that are meaningful in a population may not be applicable to an individual. A statement about reproductive risk should be included when a carrier is identified.
The interpretation should consider genes and variants with multiple disease associations, as well as a possibility of mixed modes of inheritance. For example, whereas some pathogenic variants in ABCC8 gene result in a reduced insulin secretion and hyperglycemia causing permanent neonatal diabetes mellitus, others can cause congenital hyperinsulinism and hypoglycemia. Also, although a number of pathogenic variants in ALPL hypophosphatasia are associated with an autosomal recessive disease, some variants when present in the heterozygous state are associated with an autosomal dominant disease. The possibility of manifesting heterozygotes and their associated clinical features, if such are known, as in cases of carriers of X-linked conditions (for example, cardiomyopathy in DMD carriers; primary ovarian failure in FMR1 premutation carriers) should be discussed as part of pretest counseling. Reports must be specific in designating well-known alleles that are associated with mild symptoms, for example: Asp444His variant in BTD, the Duarte allele in GALT, HBA1/HBA2 (-+/++), and the many CYP21 nonclassic mild variants. Currently, the ACMG list of secondary findings53 is not validated for reporting in the setting of general population screening.54 The transition by molecular testing laboratories to the tier-based rubric described is expected to be gradual to accommodate the changes needed to properly implement screening.
ACMG recommends:
-
The content of carrier screening panels and the corresponding ACMG tier must be described in the laboratory reports.
-
The testing approach and detectable variant types should be clearly stated.
-
Not reporting residual risk estimates because carrier frequency and the detection rate of all genes is not established.
-
Only pathogenic and likely pathogenic variants should be routinely reported.
-
Interpretation should consider genes and variants with multiple disease associations.
-
The reporting of a VUS only in the partners of identified carriers and only with consent of the patient.
Consensus question 8: What should be emphasized during pretest and post-test counseling when performing carrier screening?
Education and counseling are critical in carrier screening. Informed decision making with carrier screening is complex and ideally should be a part of preconception care to allow any of the reproductive decision-making options. Health-care professionals should inform patients of the risks, benefits, and consequences of carrier screening. After appropriate counseling that considers the patient’s needs and values, patients should be supported to make informed and autonomous decisions including the decision to not undergo carrier screening.
Carrier screening counseling should be provided by knowledgeable and appropriately trained health-care professionals and should be performed pre- and post-test. It should be noted that traditional models of genetic counseling can be both time and labor intensive. Thus, new models need to be developed and instituted for both training nongenetics providers and counseling patients. These models might include videos, chatbots, computer-based learning, or other methods of providing information to patients and assessing their understanding. Carrier screening for autosomal recessive conditions is unique when compared to other medical testing in that test results impact the likelihood of offspring of the patient having a genetic condition, while for the most part, the patient screened is healthy. However, patients with two X chromosomes, who screen positive for X-linked conditions may manifest symptoms of the condition (e.g., OTC deficiency and hemophilia) because of skewed X inactivation. This also explains why some carriers of Duchenne muscular dystrophy (DMD) experience cardiomyopathy. A subset of these patients who have a FMR1 premutation allele are at risk to develop premature ovarian insufficiency, a condition unrelated to that seen in their XY offspring (i.e., fragile X syndrome).
Pretest counseling information that all providers should be comfortable discussing:
-
Carrier screening is optional and can be performed at any time.
-
Preconception screening is recommended over prenatal screening17,19 since it may be less stressful on patients with positive screening results and it allows for the full complement of reproductive decision making. If done in pregnancy, concurrent partner testing should be offered.
-
When a reproductive partner has changed, carrier screening should be readdressed.
-
Carrier screening is not a test for all genetic conditions; in fact, considering all genetic conditions, only a minority are screened.
-
Genetic variants have likely been in one’s family for many generations.
-
Carrier screening will not identify de novo variants in the offspring.
-
Carrier screening does not replace newborn screening.
-
When Tier 1 or Tier 2 carrier screening was performed in a prior pregnancy, Tier 3 screening should be offered.
-
A carrier of an autosomal recessive condition will rarely manifest any clinical signs or symptoms of that condition.
-
Consanguineous couples have an increased risk to be carriers for the same conditions.
-
All genes and variants that cause a condition may not be known and may not be examined as part of Tier 3 or Tier 4 screening. If family history warrants, additional genes may be considered for evaluation and referral to a genetics professional should be considered. A negative test reduces the chance to have an affected child but does not eliminate the risk.
-
Laboratories should not report changes in a gene that has no or unclear association with a medical condition.
-
A VUS is a change within a gene that may or may not be associated with disease. These are not reported unless one partner is found to be a carrier of a pathogenic or likely pathogenic variant in the same gene. When this occurs the second partner should be asked to decide on whether they want this information. Ideally, this consent to return a VUS result will take place during preconception counseling.
-
In some situations, X-linked heterozygous patients will manifest signs and symptoms that are different than the condition seen in offspring (e.g., DMD, FMR1).
Counseling in specific circumstances
When screening test results are positive after sequential screening
Availability of the partner should not dictate when or if carrier screening is offered; however, the impact on interpretation of the result should be discussed as it may influence the patient’s decision making. When carrier screening is performed during an ongoing pregnancy, it is ideal to perform carrier screening on both partners simultaneously, so that screening results can be obtained in a timely manner. Carrier screening can be approached sequentially, meaning that a patient can undergo screening first, obtain results, and then a current or future reproductive partner can be screened later. When sequential screening is performed and one partner is discovered to be a carrier of an autosomal recessive or X-linked condition, that partner should undergo counseling by a knowledgeable and appropriately trained health-care professional. In specific circumstances, it may be especially appropriate to seek the assistance of a genetics professional, for example (1) when the gene or variant is known to be associated with variable expressivity, (2) when an X-linked carrier is identified, (3) when autosomal recessive carriers of gene variants that have possible phenotypic implications are identified, and (4) when a VUS is disclosed.
ACMG recommends that counseling patients include:
-
Education about the condition for which the patient tested positive.
-
Offering follow-up screening of the partner with analysis of the same gene that has the pathogenic or likely pathogenic variant as that identified in the partner.
-
Laboratory testing of the partner should include sequencing of the full gene identified in the carrier patient and not testing for a limited panel of variants.
-
In cases where there is an ongoing pregnancy and the partner declines testing or is unavailable for testing a diagnostic procedure can be offered.27
-
A plan should be made for results delivery, including whether variants of uncertain significance will be reported.
-
A negative test result in the partner does not eliminate the risk of an affected child. The remaining risk cannot be accurately quantified for most conditions, but it is reduced.
-
“False positive” results may be due to:
-
Reduced penetrance of known pathogenic and likely pathogenic variants;
-
Conflicting variant interpretation among laboratories;
-
Underreporting of outcomes in patients with same variants;
-
Imperfect in silico modeling of variant expression.
-
-
Patients should be counseled that variability of manifestations of a genetic condition is typical, even in affected individuals within the same family.
When couple is identified as being at risk
When an at-risk couple is identified, counseling by an appropriately trained health-care professional is recommended. In specific circumstances, it may be especially appropriate to seek the assistance of a genetics professional, for example (1) when the gene or variant is known to be associated with variable expressivity, and (2) when a VUS is disclosed. The counseling performed depends on when the carrier couple is identified (i.e., preconceptionally versus prenatally).
ACMG recommends that counseling patients include:
In cases of preconception identification
-
A discussion of the risks and benefits of reproductive options.
-
A discussion of in vitro fertilization with gamete donation, preimplantation genetic testing, embryo donation, adoption, and prenatal diagnosis (chorionic villi sampling or amniocentesis) followed by a decision to continue or not continue a pregnancy. This discussion includes preparation for medical care after the birth of an affected child.
-
Offering educational materials and resources that can facilitate patients in making an informed decision about their reproductive options.
-
A plan for disclosure of results.
In cases of identification during an ongoing pregnancy
-
Offering a diagnostic procedure (i.e., chorionic villi sampling or amniocentesis) as appropriate to determine whether a fetus is predicted to be affected with the condition(s) identified through carrier screening.
-
A discussion of reproductive decisions to carry a pregnancy, including preparation for possible medical care after the birth of an affected child.
-
Offering educational materials and resources that can facilitate patients in making an informed decision about their reproductive options.
-
A plan should be made for disclosure of results.
When the father cannot be screened and the patient screens positive and there is an ongoing pregnancy
It is acceptable to offer the patient a prenatal diagnostic procedure (CVS or amniocentesis) when the patient screens positive for an autosomal recessive gene and the father cannot be screened for one of the following reasons: (1) partner is unavailable for screening, (2) screening the partner would be cost prohibitive, (3) the results from the partner would not be available in time to allow for reproductive decision making, and (4) a diagnostic procedure is being performed for another reason. This option and these indications have already been established by ACMG for cystic fibrosis,27 and should be considered an option when a carrier for any other recessive gene(s) is identified. When this situation arises, counseling by an appropriately trained health-care professional is recommended. A laboratory willing to perform the testing must be identified before performing the diagnostic procedure.
ACMG recommends that counseling patients should include the following:
-
Education about the condition for which the patient tested positive.
-
A plan should be made for results delivery, including whether variants of uncertain significance will be reported.
-
Laboratory testing of the partner should include sequencing of the full gene(s) identified in the carrier patient and not testing for a limited panel of variants.
-
A diagnostic procedure should be offered when:
-
The partner is unavailable for testing;
-
The partner declines testing;
-
Testing is cost prohibitive;
-
A partner’s results would not be available in time for reproductive decision making;
-
A diagnostic procedure is already planned for another indication.
-
-
The patient should be counseled about the limitations of gene analysis in the fetus under these circumstances. The laboratory may be unable to provide definitive diagnosis if one parent’s carrier status is unknown.
CONCLUSION
This document establishes a tiered approach to carrier screening and aims to improve the implementation of carrier screening allowing diverse populations to benefit from new and emerging genomic technologies. We have listed the genes that should be offered to all patients who desire carrier screening. We realize that the genes we recommend may not adequately address those seen more frequently in some populations; therefore, family and personal history, including the pedigree and, where appropriate, physical examination, should be used to guide the need to screen selected additional genes. We expect that over time clinicians will become comfortable with the concepts, specific genes, and their associated conditions that are proposed in this document. Importantly, molecular testing laboratories are called on to adapt and innovate to keep carrier screening costs low and throughput high. It will be important that ACMG reevaluate the genes listed for screening and consider the need to modify criteria used to include and exclude genes.
The authors of this practice resource recognize that there are barriers to the implementation of Tier 3 carrier screening in clinical practice. These include the challenges imposed on health-care providers by rapidly changing genetic technologies and information, as well as insurance coverage for carrier screening of patients and partners. Another challenge is for the molecular testing laboratories to adapt new testing strategies since some of the ACMG Tier 3 genes may harbor variants that are not routinely detected by NGS only. We also recognize that the pretest counseling and delivering accurate and timely results to patients is time consuming. The information contained in this document along with that provided by ACMG, ACOG, and other professional organizations2,17,18,19,21 provides much of what needs to be known to feel comfortable offering carrier screening. This workgroup recognizes that offering a comprehensive Tier 3 panel to all is only the first step toward equity in carrier screening and clinical follow up. Working collaboratively genetics professionals are encouraged to innovate by utilizing telemedicine and online tools to overcome challenges to the workforce. Combining these with other ideas will ensure patients receive the highest level of care as genetics and genomics increases its reach into communities that, until now, were unfamiliar with their benefits. We strongly recommend that all payers provide coverage for Tier 3 carrier screening, as well as Tier 4 carrier screening in appropriate clinical circumstances such as personal/medical history or consanguinity, to ensure equitable care to all individuals including those disadvantaged by race and financial hardship.
Change history
27 August 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41436-021-01300-z
References
Grody, W. W. et al. ACMG position statement on prenatal/preconception expanded carrier screening. Genet. Med. 15, 482–483 (2013).
Committee Opinion No. 690. Carrier screening in the age of genomic medicine. Obstet. Gynecol. 129, e35–e40 (2017).
Wilson, J. M. G. & Jungner, G. Principles and Practice of Screening for Disease. (World Health Organization, Geneva, 1968).
Ram, K. T. & Klugman, S. D. Best practices: antenatal screening for common genetic conditions other than aneuploidy. Curr. Opin. Obstet. Gynecol. 22, 139–145 (2010).
Lazarin, G. A., Hawthorne, F., Collins, N. S., Platt, E. A., Evans, E. A. & Haque, I. S. Systematic classification of disease severity for evaluation of expanded carrier screening panels. PLoS One. 9, e114391 (2014).
National Tay-Sachs and Allied Diseases. https://ntsad.org/index.php/about-ntsad/our-history (2017).
Kaback, M., Lim-Steele, J., Dabholkar, D., Brown, D., Levy, N. & Zeiger, K. Tay–Sachs disease—carrier screening, prenatal diagnosis, and the molecular era. An international perspective, 1970 to 1993. The International TSD Data Collection Network. JAMA. 270, 2307–2315 (1993).
Toro, C., Shirvan, L. & Tifft, C. in GeneReviews (eds Adam, M. P. et al.) HEXA disorders. (University of Washington, Seattle, 1999).
Naik, R. P. & Haywood, C. Jr. Sickle cell trait diagnosis: clinical and social implications. Hematology Am. Soc. Hematol. Educ. Program. 2015, 160–167 (2015).
Ojodu, J., Hulihan, M. M., Pope, S. N. & Grant, A. M. Incidence of sickle cell trait—United States, 2010. MMWR Morb. Mortal. Wkly. Rep. 63, 1155–1158 (2014).
Shraga, R. et al. Evaluating genetic ancestry and self-reported ethnicity in the context of carrier screening. BMC Genet. 18, 99 (2017).
Kaseniit, K. E., Haque, I. S., Goldberg, J. D., Shulman, L. P. & Muzzey, D. Genetic ancestry analysis on >93,000 individuals undergoing expanded carrier screening reveals limitations of ethnicity-based medical guidelines. Genet. Med. 22, 1694–1702 (2020).
Westemeyer, M. et al. Clinical experience with carrier screening in a general population: support for a comprehensive pan-ethnic approach. Genet. Med. 22, 1320–1328 (2020).
Heather, J. M. & Chain, B. The sequence of sequencers: the history of sequencing DNA. Genomics. 107, 1–8 (2016).
National Human Genome Research Institute. The cost of sequencing a human genome. https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost (2020).
Shaer, O., Nov, O., Westendorf, L. & Ball, M. Communicating personal genomic information to non-experts: a new frontier for human-computer interaction. Foundations Trends Hum.–Comput. Interact. 11, 1–62 (2017).
Grody, W. W., Cutting, G. R., Klinger, K. W., Richards, C. S., Watson, M. S. & Desnick, R. J. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet. Med. 3, 149–154 (2001).
Prior, T. W. Carrier screening for spinal muscular atrophy. Genet. Med. 10, 840–842 (2008).
Committee Opinion No. 691. Carrier screening for genetic conditions. Obstet. Gynecol. 129, e41–e55 (2017).
Gregg, A. R. Message from ACMG president: overcoming disparities. Genet. Med. 22, 1758 (2020).
Edwards, J. G. et al. Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet. Gynecol. 125, 653–662 (2015).
Haddow, J. E. & Palomaki, G. E. In Human Genome Epidemiology: A Scientific Foundation for Using Genetic Information to Improve Health and Prevent Disease (eds Khoury, M. et al.). ACCE: a model process for evaluating data on emerging genetic tests. (Oxford University Press, Oxford, 2003).
Rehder, C. et al. Next generation sequencing for constitutional variants in the clinical laboratory, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. https://doi.org/10.1038/s41436-021-01139-4 (2021).
Holtzman, N. A. Promoting safe and effective genetic tests in the United States: work of the task force on genetic testing. Clin. Chem. 45, 732–738 (1999).
Burke, W. Genetic tests: clinical validity and clinical utility. Curr. Protoc. Hum. Genet. 81, 9.15.1–9.15.8 (2014).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Deignan, J. L. et al. CFTR variant testing: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 22, 1288–1295 (2020).
David, K. L. et al. Patient re-contact after revision of genomic test results: points to consider-a statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 21, 769–771 (2019).
ClinVar. https://www.ncbi.nlm.nih.gov/clinvar/ (2021).
ClinGen. https://www.clinicalgenome.org/ (2021).
Rehm, H. L. et al. ClinGen—the Clinical Genome Resource. N. Engl. J. Med. 372, 2235–2242 (2015).
Khoury, M. J. Genetics and genomics in practice: the continuum from genetic disease to genetic information in health and disease. Genet. Med. 5, 261–268 (2003).
BlueCross BlueShield Assocation. https://app.evidencestreet.com/ (2021).
Ghiossi, C. E., Goldberg, J. D., Haque, I. S., Lazarin, G. A. & Wong, K. K. Clinical utility of expanded carrier screening: reproductive behaviors of at-risk couples. J. Genet. Couns. 27, 616–625 (2018).
Johansen Taber, K. A., Beauchamp, K. A., Lazarin, G. A., Muzzey, D., Arjunan, A. & Goldberg, J. D. Clinical utility of expanded carrier screening: results-guided actionability and outcomes. Genet. Med. 21, 1041–1048 (2019).
Xi, Y. et al. Expanded carrier screening in Chinese patients seeking the help of assisted reproductive technology. Mol. Genet. Genomic Med. 8, e1340 (2020).
Cannon, J., Van Steijvoort, E., Borry, P. & Chokoshvili, D. How does carrier status for recessive disorders influence reproductive decisions? A systematic review of the literature. Expert Rev. Mol. Diagn. 19, 1117–1129 (2019).
Chokoshvili, D., Vears, D. F. & Borry, P. Growing complexity of (expanded) carrier screening: direct-to-consumer, physician-mediated, and clinic-based offers. Best Pract. Res. Clin. Obstet. Gynaecol. 44, 57–67 (2017).
Perez, A. D. & Hirschman, C. The changing racial and ethnic composition of the US population: emerging American identities. Popul. Dev. Rev. 35, 1–51 (2009).
Census, race and science. Nat. Genet. 24, 97-98 (2000).
Wilson, J. F. et al. Population genetic structure of variable drug response. Nat. Genet. 29, 265–269 (2001).
Haque, I. S., Lazarin, G. A., Kang, H. P., Evans, E. A., Goldberg, J. D. & Wapner, R. J. Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA. 316, 734–742 (2016).
Peyser, A. et al. Comparing ethnicity-based and expanded carrier screening methods at a single fertility center reveals significant differences in carrier rates and carrier couple rates. Genet. Med. 21, 1400–1406 (2019).
ACOG Committee Opinion No. 442. Preconception and prenatal carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstet. Gynecol. 114, 950 (2009).
Risch, N., Burchard, E., Ziv, E. & Tang, H. Categorization of humans in biomedical research: genes, race and disease. Genome Biol. 3, comment2007 (2002).
Brothers, K. B., Bennett, R. L. & Cho, M. K. Taking an antiracist posture in scientific publications in human genetics and genomics. Genet. Med. 23 1004–1007 (2021).
Hendrickson, B. C. et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J. Med. Genet. 46, 641–644 (2009).
Zvereff, V. V., Faruki, H., Edwards, M. & Friedman, K. J. Cystic fibrosis carrier screening in a North American population. Genet. Med. 16, 539–546 (2014).
Ben-Shachar, R., Svenson, A., Goldberg, J. D. & Muzzey, D. A data-driven evaluation of the size and content of expanded carrier screening panels. Genet. Med. 21, 1931–1939 (2019).
Guo, M. H. & Gregg, A. R. Estimating yields of prenatal carrier screening and implications for design of expanded carrier screening panels. Genet. Med. 21, 1940–1947 (2019).
Gross, S. J., Pletcher, B. A. & Monaghan, K. G. Carrier screening in individuals of Ashkenazi Jewish descent. Genet. Med. 10, 54–56 (2008).
Vankan, P. Prevalence gradients of Friedreich’s ataxia and R1b haplotype in Europe co-localize, suggesting a common Palaeolithic origin in the Franco-Cantabrian ice age refuge. J. Neurochem. 126 (Suppl 1), 11–20 (2013).
Miller, D. T. et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. https://doi.org/10.1038/s41436-021-01172-3 (2021).
Murray, M. F. et al. DNA-based screening and population health: a points to consider document for programs and sponsoring organizations from the American College of Medical Genetics and Genomics (ACMG). Genet Med. https://doi.org/10.1038/s41436-020-01082-w (2021).
OMIM. https://www.ncbi.nlm.nih.gov/omim (2021).
Santiago Borrero, P. J. et al. Genetic testing for oculocutaneous albinism type 1 and 2 and Hermansky-Pudlak syndrome type 1 and 3 mutations in Puerto Rico. J. Invest. Dermatol. 126, 85–90 (2006).
Wildenberg, S. C., Oetting, W. S., Almodovar, C., Krumwiede, M., White, J. G. & King, R. A. A gene causing Hermansky-Pudlak syndrome in a Puerto Rican population maps to chromosome 10q2. Am. J. Hum. Genet. 57, 755–765 (1995).
Witkop, C. J., Almadovar, C., Pineiro, B. & Nunez Babcock, M. Hermansky-Pudlak syndrome (HPS). An epidemiologic study. Ophthalmic Paediatr. Genet. 11, 245–250 (1990).
Ferreira, J. C., Schreiber-Agus, N., Carter, S. M., Klugman, S., Gregg, A. R. & Gross, S. J. Carrier testing for Ashkenazi Jewish disorders in the prenatal setting: navigating the genetic maze. Am. J. Obstet. Gynecol. 211, 197–204 (2014).
Bidichandani, S. I. & Delatycki, M. B. In GeneReviews (eds Adam, M. P. et al.) Friedreich ataxia. (University of Washington, Seattle, 1998).
Quinonez, S. C. & Thoene, J. G. in GeneReviews (eds Adam, M. P. et al.) Dihydrolipoamide dehydrogenase deficiency. (University of Washington, Seattle, 2014).
Piel, F. B. & Weatherall, D. J. The α-thalassemias. N. Engl. J. Med. 371, 1908–1916 (2014).
Orphanet. https://www.orpha.net/consor/cgi-bin/index.php (2021).
MedlinePlus. https://medlineplus.gov/genetics/ (2020).
Arjunan, A. et al. Evaluation and classification of severity for 176 genes on an expanded carrier screening panel. Prenat. Diagn. 40, 1246–1257 (2020).
Acknowledgements
The authors would like to thank the members of the American College of Medical Genetics and Genomics who spent their time reading this document, considering its implications and for their suggested edits.
Author information
Authors and Affiliations
Consortia
Ethics declarations
Competing interests
M.A., N.T.L., M.T.B. and E.C. are directors of molecular testing laboratories that offer carrier screening. J.S.D. is a member of the Advisory Board for Informed DNA and Medical Co-Director at Insight Medical Genetics in Chicago, which provides genetic laboratory services. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Disclaimer
This practice resource is designed primarily as an educational resource for medical geneticists and other clinicians to help them provide quality medical services. Adherence to this practice resource is completely voluntary and does not necessarily assure a successful medical outcome. This practice resource should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, the clinician should apply his or her own professional judgment to the specific clinical circumstances presented by the individual patient or specimen.
Clinicians are encouraged to document the reasons for the use of a particular procedure or test, whether or not it is in conformance with this practice resource. Clinicians also are advised to take notice of the date this practice resource was adopted, and to consider other medical and scientific information that becomes available after that date. It also would be prudent to consider whether intellectual property interests may restrict the performance of certain tests and other procedures.
The Board of Directors of the American College of Medical Genetics and Genomics approved this practice resource on 12 April 2021.
The original online version of this article was revised: Several factual errors were made on the original version of this manuscript. The authors regret the errors.
On p6: ACMG Recommends: All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening for autosomal recessive (Tables 1–5) and X-linked (Table 6) conditions. Reproductive partners of pregnant patients and those planning a pregnancy may be offered Tier 3 carrier screening for autosomal recessive conditions (Tables 1–5) when carrier screening is performed simultaneously with their pregnant partner.
First paragraph of p 10: The possibility of manifesting heterozygotes and their associated clinical features, if such are known, as in cases of carriers of X-linked conditions (for example, cardio- myopathy in DMD carriers; primary ovarian failure in FMR1 premutation carriers) should be discussed as part of pretest counseling.
On p 10 last paragraph: Carrier screening counseling should be provided by knowledgeable and appropriately trained health-care professionals and should be performed pre- and post-test. It should be noted that traditional models of genetic counseling can be both time and labor intensive. Thus, new models need to be developed and instituted for both training nongenetics providers and counseling patients. These models might include videos, chatbots, computer-based learning, or other methods of providing information to patients and assessing their understanding. Carrier screening for autosomal recessive conditions is unique when compared to other medical testing in that test results impact the likelihood of offspring of the patient having a genetic condition, while for the most part, the patient screened is healthy. However, patients with two X chromosomes, who screen positive for X-linked conditions may manifest symptoms of the condition (e.g., OTC deficiency and hemophilia) because of skewed X inactivation. This also explains why some carriers of Duchenne muscular dystrophy (DMD) or Fabry disease (GLA) experience cardiomyopathy. A subset of these patients who have a FMR1 premutation allele are at risk to develop premature ovarian insufficiency, a condition unrelated to that seen in their XY offspring (i.e., fragile X syndrome).
Bottom of p 11: When sequential screening is performed and one partner is discovered to be a carrier of an autosomal recessive or X-linked condition, that partner should undergo counseling by a knowledgeable and appropriately trained health- care professional. In specific circumstances, it may be especially appropriate to seek the assistance of a genetics professional, for example (1) when the gene or variant is known to be associated with variable expressivity, (2) when an X-linked carrier is identified, (3) when autosomal recessive carriers of gene variants that have possible phenotypic implications are identified, and (4) when a VUS is disclosed.
In addition the ESM was updated.
The original article has been corrected.
Supplementary information
Rights and permissions
About this article

Cite this article
Gregg, A.R., Aarabi, M., Klugman, S. et al. Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 23, 1793–1806 (2021). https://doi.org/10.1038/s41436-021-01203-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01203-z
This article is cited by
-
The effectiveness of expanded carrier screening based on next-generation sequencing for severe monogenic genetic diseases
Human Genomics (2024)
-
Carrier frequency estimation of pathogenic variants of autosomal recessive and X-linked recessive mendelian disorders using exome sequencing data in 1,642 Thais
BMC Medical Genomics (2024)
-
The congenital hearing phenotype in GJB2 in Queensland, Australia: V37I and mild hearing loss predominates
European Journal of Human Genetics (2024)
-
Carrier screening for present disease prevalence and recessive genetic disorder in Taiwanese population
Journal of Human Genetics (2024)
-
Prevalence of common autosomal recessive mutation carriers in women in the Southern Vietnam following the application of expanded carrier screening
Scientific Reports (2024)
