
Abstract
Purpose
Recent evolution of sequencing technologies and the development of international standards in variant interpretation have profoundly changed the diagnostic approaches in clinical genetics. As a consequence, many variants that were initially claimed to be disease-causing can be now reclassified as benign or uncertain in light of the new data available. Unfortunately, the misclassified variants are still present in the scientific literature and variant databases, greatly interfering with interpretation of diagnostic sequencing results. Despite the urgent need, large-scale efforts to update the classifications of these variants are still not sufficient.
Methods
We retrospectively analyzed 176 DYSF gene variants that were identified in dysferlinopathy patients referred to the Marseille Medical Genetics Department for diagnostic sequencing since 2001.
Results
We reclassified all variants into five-tier American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) pathogenicity classes, revealing changed pathogenicity for 17 variants. We then updated the information for the variants that have been previously published in the variant database and submitted 46 additional DYSF variants.
Conclusion
Besides direct benefit for dysferlinopathy diagnostics, our study contributes to the much needed effort to reanalyze variants from previously published cohorts and to work with curators of variant databases to update the entries for erroneously classified variants.

Similar content being viewed by others

INTRODUCTION
Dysferlinopathies are a group of autosomal recessive muscular dystrophies caused by mutations in the gene encoding dysferlin (DYSF; MIM 603009, 2p13, NM_003494.4).1,2 The two most common clinical presentations of dysferlinopathies are autosomal recessive limb-girdle muscular dystrophy 2 (LGMDR2; previously LGMD2B, MIM 253601) and Miyoshi muscular dystrophy 1 (MM; MIM 254130).3 Since description of the first pathogenic DYSF variants in 1998,1,2 more than 600 unique pathogenic or likely pathogenic variants have been reported (664 in the Leiden Open Variation Database [LOVD] [accessed 20 November 2020] and 318 variants in ClinVar [version 7 July 2020]). A significant number of these variants were identified in our diagnostic center in the Medical Genetics Department at the Timone Hospital (Marseille, France), since our laboratory was one of the first centers in Europe to offer diagnostic sequencing of all 55 exons of the DYSF gene.4,5,6
Recent evolution of sequencing technologies and bioinformatic tools has allowed accumulation of an unprecedented amount of genetic data, leading to development of large databases with variants from the general population as well as rapid expansion of databases with clinically relevant variants identified in patients with genetic diseases. These advances profoundly changed the ways variant pathogenicity is established and led to reclassification of multiple variants identified during prior diagnostic sequencing. For example, a number of variants that were previously considered pathogenic were subsequently identified in the general population at frequencies that were not compatible with pathogenicity, leading to reclassification of these variants as benign.7,8,9,10 The dramatic expansion of clinical sequencing also created a critical need for standardization of variant pathogenicity interpretation in diagnostic settings, leading to the development of a new variant classification system by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP).11 The evolution of the variant interpretation standards led to many discrepancies between the pathogenicity attributed to newly identified variants and the pathogenicity classifications of “legacy” variants identified in the earlier days of genetic sequencing. Presence of these misclassified variants in the literature and the variant databases greatly interferes with variant interpretation in the current diagnostic setting. To solve this problem for the DYSF gene, we have retrospectively evaluated the pathogenicity of 176 unique variants identified in dysferlinopathy patients referred to our center for diagnostic sequencing and updated the classification of these variants in the public database LOVD. Our results highlight the importance of variant reclassification and join the first steps of the international effort to review and reanalyze the pathogenicity classifications of variants present in the literature and public databases.
MATERIALS AND METHODS
Selection of DYSF variants for reclassification
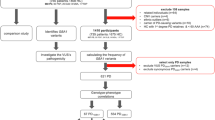
The variants described in this study were identified in patients that were referred for sequencing of the DYSF gene in our diagnostic laboratory at the Timone Children’s Hospital (Marseille, France) since 2001. The referred patients had clinical presentations highly suggestive of a primary dysferlinopathy, often with dysferlin protein deficiency identified on muscle biopsy samples (by immunohistochemistry and/or western blotting). The diagnostic approaches used during these past 20 years included single-strand conformation polymorphism (SSCP) and denaturing high performance liquid chromatography (DHPLC) with subsequent direct sequencing of identified abnormal fragments, multiplex ligation-dependent probe amplification (MLPA), quantitative polymerase chain reaction (Q-PCR), and recently a gene panel. The variants identified in dysferlinopathy patients diagnosed at our center were described in several reports4,5,6 and were the basis for the UMD-DYSF database.12 Since 2001, 572 patients and their family members have undergone diagnostic sequencing of DYSF gene in our laboratory (Fig. 1a). Of 464 suspected dysferlinopathy patients and family members referred by French neurologists, 242 patients (191 probands) carried two DYSF variants that were considered pathogenic at the moment of diagnosis. As more information about pathogenicity of DYSF variants became available over the years, classifications of certain variants have been modified. In this study, we analyzed all 176 unique DYSF variants, including those that were at one point returned as pathogenic but were then considered as benign in subsequent diagnostic returns.

(a) Five hundred seventy-two patients with a clinical presentation highly suggestive of a primary dysferlinopathy were referred for sequencing of the DYSF gene in our diagnostic laboratory at the Timone Children’s Hospital (Marseille, France) since 2001. Only patients and family members referred by French neurologists were included in this study (464 individuals). Of these, 242 patients (191 probands) carried two DYSF variants that were considered pathogenic at the time of diagnosis. The set of analyzed variants consisted of 176 unique DYSF variants, including those that were at one point returned as pathogenic but were then considered as benign in subsequent diagnostic returns. (b) The vast majority of 176 variants were returned before the five-tier American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) classification system was adopted in our laboratory. Left side of the panel shows the four types of variants analyzed: 35 nonsense variants, 53 indels, 28 variants affecting splicing, and 60 missense variants. Right side of the panel shows the numbers of variants reclassified into five ACMG/AMP pathogenic classes (pathogenic, likely pathogenic, variant of uncertain significance [VUS], likely benign, and benign), grouped by variant type. Of 176 variants, pathogenicity classification of 17 variants was downgraded to VUS, likely benign, or benign (overall concordance with original classification of 90%). Missense variant group had the largest proportion of variants with downgraded pathogenicity (16 of 60). One in-frame indel was also downgraded to VUS. The variants analyzed in this study are listed in the Supplementary Table S1. Detailed information about the lines of evidence used for variant classification is provided in the Supplementary Tables S2 and S3.
Variant classification and data analysis are described in the Supplementary Methods.
RESULTS
To address the critical issue of outdated pathogenicity classifications in public databases, we performed a retrospective analysis of 176 DYSF variants identified in the large number of patients referred to our center for diagnostic sequencing of the DYSF gene since 2001. Our laboratory was one of the first diagnostic centers in Europe to offer analysis of all 55 exons of the DYSF gene, providing this diagnostic test to 572 suspected dysferlinopathy patients and their family members over the last 20 years. We focused on families referred for sequencing by French neurologists and curated 176 unique DYSF variants identified in 242 patients (191 probands) carrying two DYSF variants that were considered pathogenic at the moment of diagnosis (Fig. 1a). It is important to note that certain variants were initially reported as pathogenic, but later were reconsidered as benign when more information about population frequencies or in trans/cis occurrences became available. We decided to include this type of variant in our analysis, since certain of them are still listed as pathogenic in public databases and could potentially influence clinical interpretations of these variants in the future. The variants curated in this study are listed in Supplementary Table S1, showing each variant’s location, type and the final pathogenicity group as well as the concordance of our classification with classifications in ClinVar and LOVD at the time of the study. Supplementary Table S2 provides the detailed information about the lines of evidence and the ACMG/AMP codes used to classify each variant.
The vast majority of variants in our study were returned before the five-tier ACMG/AMP classification was adopted in our laboratory in 2016. Figure 1b shows the number of variants reclassified into five ACMG/AMP pathogenicity classes, grouped by type of the variant (35 nonsense variants, 53 indels, 28 variants affecting splicing, and 60 missense variants). Nonsense and indel variants had the largest proportion of variants assigned to the pathogenic group (34/35 and 46/53 respectively). The missense variant group had the largest proportion of variants with downgraded pathogenicity (16 of 60). Of 176 variants, 17 variants were reclassified as variants of uncertain significance (VUS), likely benign, or benign (overall concordance with original classification of 90%). LOVD entries for all variants with changes in classification were updated. Supplementary Table S3 provides detailed information used for assigning these variants into the pathogenicity classes and lists the types of evidence that were missing to classify a given variant to a pathogenic or likely pathogenic category. The major reasons for assigning variants to VUS/likely benign/benign categories were lack of other patients carrying the analyzed variant (7/17), absence of the second pathogenic/likely pathogenic variant (6/17), or association with two pathogenic/likely pathogenic variants (4/17). As a consequence, the PM3 code was not attributed or attributed at a “supporting” level for 12 and 5 variants respectively and the PP4 code was not attributed for 12 variants. Seven variants in this group had elevated allele frequencies (BA1, BS1, or lack of PM2) and six variants were not predicted as deleterious by in silico algorithms (no PP3). Finally, three variants in this group were associated in cis with a pathogenic or a likely pathogenic variant (BP2).
Of the variants with confirmed pathogenicity, 22 had at least one entry listed as uncertain in LOVD or ClinVar at the time of the study (Table S1). In most cases, additional previously unreported patient or family segregation data allowed to establish firmer pathogenicity, thus decreasing the possibility of misdiagnosis for future patients with these variants. Variant locations along the DYSF protein and its functional domains are shown in Fig. 2. Forty-three clinically relevant (pathogenic and likely pathogenic) missense variants identified in patients referred to the Marseille center for DYSF sequencing are shown in the top panel (gray). One hundred fifteen clinically relevant indel, nonsense, and splice variants are shown in the lower panel (purple). Loss-of-function variants (purple) are distributed homogeneously along the protein length, while missense variants (gray) are more often found in C2 and Dysf domains.

Forty-three clinically relevant (pathogenic and likely pathogenic) missense variants identified in dysferlinopathy patients referred for sequencing to the Department of Medical Genetics in Marseille are shown in the top panel (gray). One hundred fifteen clinically relevant indel, nonsense, splice variants identified in this set of samples are shown in the lower panel (purple). Exon–intron junctions of the DYSF gene as well as the functional domains of dysferlin are visualized. The dysferlin domain nomenclature is shown according to Sula et al.20 The variants analyzed in this study are listed in the Supplementary Table S1. Detailed information about the lines of evidence used for variant classification is provided in the Supplementary Table S2.
Forty variants identified in dysferlinopathy patients in our center were novel, as they were not present in LOVD or in ClinVar. We submitted all variants that have not been previously reported by our group and updated the previously reported variants4,5,6 in LOVD.
DISCUSSION
Since the ACMG/AMP classification system became widely accepted, a certain number of studies have described retrospective reanalysis of variants in various genetic conditions. Reinterpretation of variants can be approached from two perspectives: variant-based and patient-based. In the first approach, variants are reinterpreted based on publicly available data, such as population frequency or recent publications. Large-scale studies of this type can be undertaken by any group without need to access patient-related information.13,14 In comparison, patient-based variant reclassification takes into account patient and family related data, such as segregation and phenotypic data, in addition to the updated variant frequency and literature information. This reclassification has to be done by the same group that published the original cohort, thus making this type of study more difficult to perform and less frequent.15,16,17 In our study, we retrospectively analyzed variants from a large series of dysferlinopathy patients, allowing reclassification of 17 variants.
Of 176 variants analyzed in this study, 159 retained pathogenic or likely pathogenic classification after the re-evaluation. Several aspects of our study explain this relatively low concordance (90%). First, we included in the analysis certain variants that were identified as pathogenic at the very beginning of diagnostic sequencing of DYSF when the knowledge about this gene and its variants was still very limited. Even though these variants were later reinterpreted as benign or uncertain and were no longer returned to patients as pathogenic, we thought that it was important to formally reclassify these variants and to correct the “pathogenic” classification in public databases such as LOVD. Second, earlier pathogenicity classifications were attributing much higher weight to predictions by in silico algorithms, in particular for the interpretation of missense variants. In the current ACMG/AMP classifications, pathogenicity predictions are used as only a supporting line of evidence (evidence code PP3).11 Third, dysferlinopathies are much rarer than many other inherited disorders, making it difficult to interpret the pathogenicity of a variant found in only one or several patients. Indeed, a recent study reinterpreting hereditary cancer variants in such frequently mutated genes as BRCA1 had a higher concordance for pathogenic variants (99.3%).18 Fourth, interpretation of loss-of-function variants is much more straightforward than that of missense variants. As seen from Fig. 1b and S1, most of the discordance came from missense variants. Indeed, no protein truncating variants had downgraded classifications, while the classifications of 16 missense variants and one small in-frame indel were changed to VUS, likely benign, or benign (see Supplementary Table S3 for more details). Since missense variants in the DYSF gene are a common cause of dysferlinopathy, the concordance between reinterpretations of DYSF variant pathogenicity is expected to be lower compared with the concordance between reinterpretations of pathogenic variants in a gene where only loss-of-function variants cause disease.
Despite recent advances aimed at standardizing the classification of variants identified by diagnostic sequencing, there are still many examples of erroneous pathogenicity classification in the scientific literature and public databases of pathogenic variants.7,8,10 The variant interpretation in skeletal muscle disorders could be even more difficult due to reduced penetrance of certain variants as well as possible digenic or multigenic modes of inheritance. Ongoing international curation efforts, such as the ones led by ClinGen, aim to establish gene-specific interpretation guidelines and to curate a certain number of variants.19 However, the outdated variant classifications that are currently found in the variant databases and the literature are not likely to be completely corrected by these efforts. Our study retrospectively analyzes DYSF variants identified, published, and submitted to variant databases during 20 years of diagnostic sequencing at the Medical Genetics Department in Marseille, France.4,5,6 Besides direct benefit for diagnostics of dysferlinopathies, our study is an example of much needed effort to re-evaluate variant interpretations in the previously published cohorts and to work with curators of variant databases to update the information for the erroneously classified variants.
Data availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files. Forty novel variants were submitted to LOVD-DYSF database (www.lovd.nl/DYSF). Overall, 46 new entries were made to LOVD-DYSF and 130 previous entries were updated.
References
Liu, J. et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 20, 31–36, https://doi.org/10.1038/1682 (1998).
Bashir, R. et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 20, 37–42, https://doi.org/10.1038/1689 (1998).
Aoki, M. In GeneReviews (eds Adam, M. P. et al.) Dysferlinopathy (University of Washington, Seattle, 1993). http://www.ncbi.nlm.nih.gov/books/NBK1303/.
Nguyen, K. et al. Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum. Mutat. 26, 165, https://doi.org/10.1002/humu.9355 (2005).
Nguyen, K. et al. Phenotypic study in 40 patients with dysferlin gene mutations: high frequency of atypical phenotypes. Arch. Neurol. 64, 1176–1182, https://doi.org/10.1001/archneur.64.8.1176 (2007).
Krahn, M. et al. Analysis of the DYSF mutational spectrum in a large cohort of patients. Hum. Mutat. 30, E345–E375, https://doi.org/10.1002/humu.20910 (2009).
Piton, A., Redin, C. & Mandel, J.-L. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 93, 368–383, https://doi.org/10.1016/j.ajhg.2013.06.013 (2013).
Bell, C. J. et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 3, 65ra4, https://doi.org/10.1126/scitranslmed.3001756 (2011).
Whiffin, N. et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 19, 1151–1158, https://doi.org/10.1038/gim.2017.26 (2017).
Di Fruscio, G., Garofalo, A., Mutarelli, M., Savarese, M. & Nigro, V. Are all the previously reported genetic variants in limb girdle muscular dystrophy genes pathogenic? Eur. J. Hum. Genet. 24, 73–77, https://doi.org/10.1038/ejhg.2015.76 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424, https://doi.org/10.1038/gim.2015.30 (2015).
Blandin, G. et al. UMD-DYSF, a novel locus specific database for the compilation and interactive analysis of mutations in the dysferlin gene. Hum. Mutat. 33, E2317–E2331, https://doi.org/10.1002/humu.22015 (2012).
Niestroj, L.-M. et al. Guideline-based and bioinformatic reassessment of lesion-associated gene and variant pathogenicity in focal human epilepsies. Epilepsia. 59, 2145–2152, https://doi.org/10.1111/epi.14579 (2018).
Harrison, S. M. et al. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet. Med. 19, 1096–1104, https://doi.org/10.1038/gim.2017.14 (2017).
Cerino, M. et al. Refining NGS diagnosis of muscular disorders. J. Neurol. Neurosurg. Psychiatry. https://doi.org/10.1136/jnnp-2018-319254 (2020).
Krenn, M. et al. Genotype-guided diagnostic reassessment after exome sequencing in neuromuscular disorders: experiences with a two-step approach. Eur. J. Neurol. 27, 51–61, https://doi.org/10.1111/ene.14033 (2020).
Tsai, G. J. et al. Outcomes of 92 patient-driven family studies for reclassification of variants of uncertain significance. Genet. Med. 21, 1435–1442, https://doi.org/10.1038/s41436-018-0335-7 (2019).
Mersch, J. et al. Prevalence of variant reclassification following hereditary cancer genetic testing. JAMA. 320, 1266–1274, https://doi.org/10.1001/jama.2018.13152 (2018).
Rivera-Muñoz, E. A. et al. ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum. Mutat. 39, 1614–1622, https://doi.org/10.1002/humu.23645 (2018).
Sula, A., Cole, A. R., Yeats, C., Orengo, C. & Keep, N. H. Crystal structures of the human Dysferlin inner DysF domain. BMC Struct. Biol. 14, 3, https://doi.org/10.1186/1472-6807-14-3 (2014).
Acknowledgements
We thank the neurologists in France and beyond for referring patients to our diagnostic center. We also thank the entire team of the Laboratory of Molecular Genetics at the Timone Hospital, Marseille, for their assistance in establishing the diagnostic sequencing of the DYSF gene as well as for their help with this analysis over the years. This study was supported by Genetics Institute for Patients, Therapies Innovation & Science (GIPTIS), AFM-TELETHON, and Assistance Publique–Hôpitaux de Marseille (AP-HM).
Author information
Authors and Affiliations
Contributions
T.C. had a major role in the analysis of data and participated in writing of the manuscript; V.B. and C.P. participated in data acquisition; M.B., M.C., F.R., N.B.-P., K.N., and N.L. revised the manuscript for content; M.K. and S.G. had major roles in the conception of the study, data interpretation, and manuscript writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declaration
The data on DYSF variants do not include any identifying information. These data correspond to retrospective analysis that does not require ethics committee approval at our institution. The appropriate consent was collected from each patient at the moment of variant identification as part of the standard procedure during a diagnostic genetic testing in our laboratory.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
co-last authors: Svetlana Gorokhova, Martin Krahn.
Supplementary information
Rights and permissions
About this article

Cite this article
Charnay, T., Blanck, V., Cerino, M. et al. Retrospective analysis and reclassification of DYSF variants in a large French series of dysferlinopathy patients. Genet Med 23, 1574–1577 (2021). https://doi.org/10.1038/s41436-021-01164-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01164-3
