
Abstract
Purpose
We characterize the clinical and molecular phenotypes of six unrelated individuals with intellectual disability and autism spectrum disorder who carry heterozygous missense variants of the PRKAR1B gene, which encodes the R1β subunit of the cyclic AMP-dependent protein kinase A (PKA).
Methods
Variants of PRKAR1B were identified by single- or trio-exome analysis. We contacted the families and physicians of the six individuals to collect phenotypic information, performed in vitro analyses of the identified PRKAR1B-variants, and investigated PRKAR1B expression during embryonic development.
Results
Recent studies of large patient cohorts with neurodevelopmental disorders found significant enrichment of de novo missense variants in PRKAR1B. In our cohort, de novo origin of the PRKAR1B variants could be confirmed in five of six individuals, and four carried the same heterozygous de novo variant c.1003C>T (p.Arg335Trp; NM_001164760). Global developmental delay, autism spectrum disorder, and apraxia/dyspraxia have been reported in all six, and reduced pain sensitivity was found in three individuals carrying the c.1003C>T variant. PRKAR1B expression in the brain was demonstrated during human embryonal development. Additionally, in vitro analyses revealed altered basal PKA activity in cells transfected with variant-harboring PRKAR1B expression constructs.
Conclusion
Our study provides strong evidence for a PRKAR1B-related neurodevelopmental disorder.
Similar content being viewed by others


INTRODUCTION
The gene PRKAR1B (Protein Kinase cAMP-Dependent Type I Regulatory Subunit Beta) encodes a regulatory subunit of the cyclic AMP-dependent protein kinase A protein complex (PKA), which is a nearly universal cellular component in eukaryotes.1 PKA is a heterotetramer of two regulatory (R) and two catalytic (C) subunits, which, upon activation of PKA by cAMP, phosphorylates serine or threonine residues of different target proteins. In humans, the genes PRKAR1A, PRKAR1B, PRKAR2A, and PRKAR2B encode the regulatory subunits RIα, RIβ, RIIα, and RIIβ, while the genes PRKCA and PRKCB give rise to a total of six principal catalytic subunit isoforms: Cα1, Cα2, and Cβ1–4. Cell type–specific expression of different subunits changes the composition and thereby intracellular localization and substrate specificity of PKA isoforms.2 R subunits serve as cAMP receptors and facilitate the spatial localization of PKA within the cell by binding different A-Kinase anchoring proteins (AKAPs).3 The subunit RIβ is primarily expressed in the brain,4,5 with the highest levels of expression in the cerebral cortex and hypothalamus.6
The first functional study of R1β involving R1β-deficient mice was completed 25 years ago, with mice deficient of the murine ortholog of R1β showing altered hippocampal long-term depression and depotentiation.7 Downregulation of R1β in murine hippocampal cultures was found to reduce the phosphorylation of CREB,8 a transcription factor implicated in long-term memory formation.9 Furthermore, R1β-deficient mice showed diminished nociceptive pain and inflammation in the setting of persistent tissue injury, although the reaction to acute nociceptive stimuli was unaffected.10
A missense variant in PRKAR1B has been associated with a rare hereditary neurodegenerative disorder in humans with R1β-positive inclusions in affected neurons,11,12 but there is also mounting evidence for a role of PRKAR1B in neurodevelopmental disorders (NDDs): statistical analyses of a cumulative data set of 10,927 cases derived from several NDD patient cohorts found recurrent sites (defined as “de novo missense variants of the same amino acid in two or more unrelated cases”) in PRKAR1B, among other potential NDD candidate genes.13 An analysis of expression profiles and protein–protein interaction14 of 253 NDD candidate genes based on the same data set positioned PRKAR1B among a network of genes related to c-Jun N-terminal kinase and mitogen-activated protein kinase cascades, which contained several previously identified NDD candidate genes.15 Another recent study found a significant enrichment of de novo missense variants in PRKAR1B in a large sample of 31,058 trio exomes of children with developmental disorders and their unaffected parents.16
PRKAR1B is therefore a promising candidate gene for NDDs, including autism spectrum disorder (ASD),17 although no clear Mendelian disease association has been established to date. We now report six unrelated individuals with variants of PRKAR1B, who share similar features indicative of a neurodevelopmental disorder.
MATERIALS AND METHODS
Exome sequencing
Trio-based exome sequencing was performed for individuals 1, 3, 4, 5, and 6. Proband-only based exome sequencing was performed for individual 2, as parental samples were not available. Individuals 1, 2, 3, 5, and 6 were enrolled through GeneDx. Individual 4 was enrolled through the University of California–Los Angeles (UCLA) Clinical Site of the Undiagnosed Diseases Network (UDN). Following informed consent, a comprehensive chart review of medical records was performed.
DNA constructs and cell culture
The wild type (WT) (NM_001164760.2) and three variant (Glu196Lys, p.Gln167Leu, and p.Arg335Trp) PRKAR1B sequences were introduced into pVenus-PRKAR1B vector according to the method described elsewhere.18,19,20 The vector pCerulean-PRKACA was also created according to the method described elsewhere.19
PKA enzymatic activity assay
HEK293 cells were transfected with the three previously described constructs (Glu196Lys, p.Gln167Leu, and p.Arg335Trp) using Lipofectamine 3000 (Invitrogen) and were harvested 24 hours post-transfection. Cells were lysed in freshly prepared lysis buffer (10 mM Tris-HCl (pH 7.5), 1 mM EDTA, and 1 mM dithiothreitol with 0.5 mM PMSF and protease inhibitor cocktail I (1:100; EMD Biosciences, La Jolla, CA). BCA assays were performed as per manufacturer’s protocol to determine the total protein concentrations of samples (Pierce). Samples were diluted to 0.5 µg/µL and 10 µL of lysate was used for each reaction. PKA enzymatic assays were performed by kemptide assay, using 25 µM kemptide (Leu-Arg-Arg-Ala-Ser-Leu-Gly), as previously described with and without cAMP (5 µM).21 All reactions for basal and cAMP-stimulated (total) PKA activity were carried out in duplicate. Additionally, activity values for replicate reactions that were incubated in the presence of PKI (5 nM) were subtracted from activity values to account for nonspecific kinase activity.
Fluorescence resonance energy transfer (FRET) by acceptor photobleaching
HEK293 cells (ATCC) were seeded onto 12-well plates and left overnight to recover. The HEK293 cells were transfected with R1β-Venus and Cα-Cerulean vectors (1 μg each) and 9 hours later the cells were placed in low-serum medium (0.5% FBS) for 12 hours before stimulations. Experiments were performed on the confocal microscope Zeiss LSM 880 Airyscan as described previously (Zeiss, NY).22 Cα-Cerulean was imaged with the 405-nm laser, R1β-Venus with the 514-nm laser. Using a custom region of interest (ROI), R1β-Venus in one cell was bleached with the 514-nm laser at 100% transmission until the overall intensity dropped to between 80% and 50% of prebleach values. The R1β-Cα interaction was calculated by the difference in Cerulean intensity pre- versus postbleaching. In each experiment, 10–15 bleaches on different cells were performed. Results shown are the combined results of three separate experiments.
RNAscope in situ hybridization assay
Embryos were collected by the Human Developmental Biology Resource (https://www.hdbr.org) with ethics approval and following appropriate consent. First, 8-μm tissue sections were taken through the brain and the slides were baked for 1 hour at 60 °C before the paraffin was removed in xylene and the sections were dehydrated in two changes of 100% ethanol. Then, 1× target retrieval was performed by heating the sections for 20 minutes at 95 °C, followed by protease treatment for 15 minutes at 40 °C. An RNAscope PRKAR1β probe (ID 861041-C2) was hybridized to the tissue for 2 hours at 40 °C followed by multiple signal amplification steps. Probe hybridization was detected using Fast Red and the sections counterstained with 50% hematoxylin for 30 seconds at room temperature.
Statistics
PKA enzymatic assay
For the PKA enzymatic activity assay, data were normally distributed as determined by Shapiro–Wilk test. One-way analysis of variance (ANOVA) was performed for both the basal and total PKA activity data sets and Bonferroni multiple comparison test was used for basal activity data that produced a significant ANOVA statistic.
FRET experiment
For the FRET experiment, data normality was assessed by Shapiro–Wilk test and the appropriate statistical test was used. For the FRET by acceptor photobleaching experiment, data were not normally distributed and Mann–Whitney test was used. Statistical significance was set at P < 0.05 and analyses were carried out using the GraphPadPrism 6 (GraphPad®) software.
Comparison of RNA-Seq expression levels
For comparing RNA-Seq expression levels of different data sets from different sources23,24,25 (e.g., different experimental setup and analysis software), a normalization of the provided expression scores was necessary. We derived the distribution of expression scores over all genes and calculated the percentile of each gene of interest within this distribution. This was done individually for each data set. As a result, each scoring is limited to the range between 0% and 100%, where 0% reflects the respective gene with the lowest level of expression, and 100% the gene with the highest level of expression within the corresponding data set. The median expression level of each data set is set to 50%.
Since every source provides a single measurement of the expression levels of a certain gene, we used a bootstrapping algorithm with a sample size of 1,000 and 5 repetitions, for the derivation of the uncertainties of the resulting percentiles (error bars).
RESULTS
We report six individuals with variants in the PRKAR1B gene (five males, one female; mean age 8.83 years, age range 3–16 as of June 2020). All six individuals were diagnosed with ASD by an expert physician, following DSM-V criteria. Global developmental delay (GDD) was reported in all, and congenital hypotonia was reported in three individuals. All individuals had neurologic anomalies, predominantly disorders of movement. These included dyspraxia/apraxia and clumsiness in all, tremor and dystonia in one, and involuntary movements (eye twitching) in another individual. High pain tolerance was reported by the parents of three individuals, with one individual occasionally harming himself without noticing, and another one sometimes biting himself, when frustrated, to the point of breaking the skin. Only individual 2, whose mother was also reported to have a seizure disorder, manifested seizures.
While speech delay was reported in all individuals, speech regression has been reported in two of them: one individual lost the ability to use two-word phrases, while another one, who previously acquired an active vocabulary of ~40 words, lost most of it by age two, and became almost nonverbal from the age of five years onward. Behavioral abnormalities included autistic features like arm/hand flapping, repetitive, and sensory-seeking behavior. Attention deficit hyperactivity disorder (ADHD) was clinically diagnosed in four individuals and suspected by the parents of a fifth. Bouts of aggression were reported in three. Obesity (body mass index [BMI] > 30 kg/m2) was present in one individual, while another one had a BMI in the borderline obese range (BMI 29.9 kg/m2). Obsession with food was also reported in one of the other individuals. A compilation of phenotypic features of each individual can be found in Table 1. No consistent pattern of major malformations or physical anomalies was reported in our small cohort, although one individual had microcephaly and another one had plagiocephaly. The evaluation of facial photographs of individuals 1, 4, 5, and 6 did not reveal any consistent facial dysmorphisms other than upslanting palpebral fissures, which were seen in individuals 1, 2, 4, and 5 (Fig. 1a, Table 1).

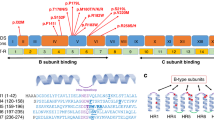
(a) Top left: individual 1 at the age of 3 years. Top right: individual 4 the age of 7 years. Bottom left: individual 5 the age of 7 years. Bottom right: individual 6 at the age of 3 years. (b) Distribution of the observed variants within the PRKAR1B gene. Exons are shown as boxes, introns as a blue line (introns are not to scale). Light blue color indicates protein-coding sequence. (c) Mutated amino acid (AA) positions within the R1β protein and number of affected individuals. A color shift to red indicates a higher degree of intolerance towards AA variation throughout evolution; according to the respective position’s evolutionary trace (ET) score (see Supplemental Methods for further details).
De novo origin of the respective PRKAR1B variants has been confirmed in five individuals, and the parents of individual 2 were not available for testing. Four individuals carry the same variant c.1003C>T (p.Arg335Trp), while the other two carry the missense variants: c.586G>A (p.Glu196Lys) and c.500_501inv (p.Gln167Leu) (NM_001164760), respectively. A 3D model of the human R1β subunit structure26 is shown in Fig. 1b, highlighting the affected amino acid (AA) residues, which are situated within annotated nucleotide binding regions according to the UniProt database. All reported variants are absent from presumably healthy controls in public databases (gnomAD v2 and v3).27 Combined Annotation Dependent Depletion (CADD v1.6) scores28 calculated for the single-nucleotide variants c.1003C>T and c.586G>A were 26.4 and 24.1, respectively, ranking these variants among the most deleterious 1% of substitutions in the human genome as predicted by the CADD software. The variant c.500_501inv is situated close to the 3’ border of exon 5, and predicted functional impacts on splicing vary between the different prediction tools embedded in the Alamut VisualTM variant analysis software (v2.15), ranging from −1.6% (NNSPLICE) to −46.8% (GeneSplicer). To further estimate the functional impact, we calculated evolutionary action (EA) scores29 of the three variants, which predict functional impacts of AA substitutions by taking into account the phylogenetic distances of AA changes throughout the evolutionary history of the protein, as well as the compatibility of new substitutions with the typical substitutions observed between homologous sequences. The multiple sequence alignment of 106 PRKAR1B orthologues and paralogues used in this calculation is shown in Fig. S1. The EA scores are normalized on a scale from 0 (predicted WT protein activity) to 100 (predicted loss of protein activity), with the value representing the percentage of all possible AA substitutions in the protein that have less impact. The p.Glu196Lys, p.Gln167Leu, and p.Arg335Trp AA substitutions had EA scores of 30, 84, and 87, respectively, indicating strong functional impact for the last two, mostly due to the evolutionary pressure to preserve the Gln167 and Arg335 residues. The relatively low EA score of the p.Glu196Lys substitution is due to higher variation of this AA residue across species (67.6% Glu, 7.6% Asp, 5.7% Pro, 4.8% Ala, 4.8% Asn, 2.9% Gln, 1.9% Thr, 1.9% His, 1% Arg, and 1% Val), which however still hold a negative or neutral charge, versus the substitution of glutamic acid by positively charged lysine in the affected patient, leading to an actual alteration in charge for the respective residue.
While in silico analyses suggested a deleterious effect of the three variants on R1β protein function, analyzing the effect of mutant PRKAR1B on PKA kinase in vitro could potentially validate this prediction. We performed functional studies using pVenus-PRKAR1B expression constructs for WT PRKAR1B, as well as the three variants p.Glu196Lys, p.Gln167Leu, and p.Arg335Trp. An enzymatic activity assay of lysates of HEK293 cells transfected with WT and variant-harboring constructs revealed significantly decreased basal PKA enzymatic activity in lysates of cells transfected with each of the three variant-harboring constructs (ANOVA, p = 0.0012). While total, or cAMP-stimulated PKA activity was not significantly different in cells transfected with WT constructs compared with those transfected with the three variant-harboring constructs (ANOVA, p = 0.079), the total PKA activity in lysates of cells transfected with the p.R335W construct tended to be lower (p = 0.060) (Fig. 2a). To explore whether the differences of basal PKA activity might be caused by impaired integration of mutant R1β into the PKA complex, fluorescence resonance energy transfer (FRET) studies were performed to detect minute positional differences of mutant and WT R1β proteins relative to the main PKA complex. In these studies, the three different variant-harboring R1β proteins appeared to bind less tightly to the main PKA catalytic subunit Cα at an approximately equal rate compared with WT R1β; The differences in the normalized energy transfer between the WT R1β-Cα interaction and the interactions of the three variant-harboring R1β proteins with Cα were small, yet significant, for all three p.Gln167Leu, p.Glu196Lys, and p.Arg335Trp variants (Fig. 2b).

(a) PKA enzymatic activity assay: basal and total PKA enzymatic activity in lysates of HEK293 cells transfected with PRKAR1B expression constructs (wild type [WT], p.Q167L, p.E196K and p.R335W [p.Gln167Leu, p.Glu196Lys and p.Arg335Trp]). One-way analysis of variance (ANOVA) was performed for both basal and total PKA activity data sets; a Bonferroni multiple comparison test was used for basal activity data that produced a significant ANOVA statistic. (b) Fluorescence resonance energy transfer (FRET) in HEK293 cells transfected with R1β-Venus (WT, p.Q167L, p.E196K and p.R335W) and Cα-Cerulean vectors. A Mann–Whitney U-test was used to check for statistical significance as data were not normally distributed.
As a potential NDD disease gene, PRKAR1B would be expected to be expressed in the brain during embryonal development. PRKAR1B expression is seen in different embryonic cell types including neural progenitor cells (NPCs) and neural crest cells (NCCs) (Fig. 3a).23,24,25 Additionally, an in situ hybridization assay to detect PRKAR1B messenger RNA (mRNA) (RNAscope) revealed high PRKAR1B expression in the pituitary, diencephalon, mesencephalon, and hypothalamus in human embryos at Carnegie stage 22 (Fig. 3b).

(a) Normalized expression levels of different PRKAR genes and a selection of reporter genes in embryonic stem cells (ESCs), neural progenitor cells (NPCs), and neural crest cells (NCCs), based on RNA-Seq data from different sources.23,24,25 For each individual set of expression data (ESC, NPC, and NCC), 0% reflects the gene with the lowest, and 100% the gene with the highest level of expression. The median expression level of each data set is 50% (dashed line). Genes scoring higher than 50% can be considered to be more highly expressed than the majority of genes in their respective data set. (b) Upper row: sagittal section of a human embryo at Carnegie stage 22 and corresponding 3D model of the embryonic brain (yellow: mesencephalon; green: subpallium; light blue: diencephalon; purple: hypothalamus; pink: rhombencephalon). A RNAscope PRKAR1β probe has been used to hybridize PRKAR1B messenger RNA (mRNA) (red). The section has been counterstained with hematoxylin (blue). A corresponding positive and negative control is shown in Fig. S2. Lower row: magnified sections show PRKAR1B expression in the pituitary, diencephalon, mesencephalon, and hypothalamus.
DISCUSSION
This study presents a systematic characterization of a NDD associated with missense variants in PRKAR1B. In addition to the observed enrichment of de novo missense variants of this gene in two large, independent cohorts of individuals with NDDs,15,16 the recurrent finding of the de novo variant c.1003C>T in phenotypically similar individuals strongly suggests that this variant is causative for the observed phenotype (see Supplemental Note 1), and indicates a potential mutational hotspot in the Arg335 residue. The observation of altered basal PKA activity in HEK293 cells transfected with variant-harboring constructs supports the hypothesis of a deleterious effect of the reported PRKAR1B variants on PKA function. It is important to note, however, that a decreased basal PKA enzymatic activity has been associated with a tighter binding of R1β to the Cα subunit in past studies.20 It is therefore possible that the small differences between the WT and variant-harboring R1β subunits identified by FRET do not actually cause a weaker R1β-Cα interaction, but may rather point toward another, yet unknown, molecular disease mechanism.
While expression of PRKAR1B in the adult brain had been previously shown,5 the high expression of the gene in the embryonic brain at CS22 points toward an early role of PRKAR1B in the development of the brain, which would be consistent with the concept of a PRKAR1B-associated neurodevelopmental disorder.
The clinical features of our cohort seem to approximate some aspects of the phenotype of R1β-deficient mice, such as increased pain tolerance,10 which was reported in three patients carrying the c.1003C>T variant. Furthermore, defects in hippocampal homosynaptic long-term depotentiation and low frequency stimulus-induced synaptic depression reported in mice,7 if also present in humans, may influence synaptic plasticity and thereby potentially impair learning and other cognitive functions. On a cellular level, cognitive abnormalities may be caused by diminished PKA-mediated phosphorylation of CREB in neurons. As the cAMP/PKA/CREB cascade is instrumental for the transcription of memory-associated genes and long-term memory formation,9,30 further studies might demonstrate functional impacts of PRKAR1B variants by measuring CREBP phosphorylation and expression of target genes in human IPSC-derived neurons from affected individuals and healthy controls.
We propose a PRKAR1B-associated NDD with GDD, ASD, neurologic anomalies, and cognitive impairment (no formal IQ scores have yet been obtained from the reported individuals) as principal features. Future research should focus on better understanding the functional consequences of PRKAR1B variants, as well as the recruitment and clinical characterization of more individuals carrying de novo PRKAR1B variants.
Web resources
The Genome Aggregation Database (gnomAD). https://gnomad.broadinstitute.org. Combined Annotation Dependent Depletion (CADD). https://cadd.gs.washington.edu/. UniProt. https://www.uniprot.org. Human Developmental Biology Resource. https://www.hdbr.org.
Data availability
All data supporting the conclusions of this study are presented within the article and its supplement. The authors are willing to share materials, data sets, and protocols utilized in the acquisition of data presented in this publication with other researchers upon request.
References
Taylor, S. S., Keshwani, M. M., Steichen, J. M. & Kornev, A. P. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2517–2528 (2012).
Soberg, K. & Skalhegg, B. S. The molecular basis for specificity at the level of the protein kinase a catalytic subunit. Front. Endocrinol. (Lausanne). 9, 538 (2018).
Autenrieth, K., Bendzunas, N. G., Bertinetti, D. D., FWPD, Herberg & EJPD, Kennedy Defining a-kinase anchoring protein (AKAP) specificity for the protein kinase A subunit RI (PKA-RI). Chembiochem. 17, 693–697 (2016).
Cadd, G. & McKnight, G. S. Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron. 3, 71–79 (1989).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science. 347, 1260419 (2015).
Sjostedt, E. et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science. 367, eaay5947 (2020).
Brandon, E. P. et al. Hippocampal long-term depression and depotentiation are defective in mice carrying a targeted disruption of the gene encoding the RI beta subunit of cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 92, 8851–8855 (1995).
Ilouz, R. et al. Isoform-specific subcellular localization and function of protein kinase A identified by mosaic imaging of mouse brain. Elife. 6, e17681 (2017).
Kandel, E. R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain. 5, 14 (2012).
Malmberg, A. B., Brandon, E. P., Idzerda, R. L., Liu, H., McKnight, G. S. & Basbaum, A. I. Diminished inflammation and nociceptive pain with preservation of neuropathic pain in mice with a targeted mutation of the type I regulatory subunit of cAMP-dependent protein kinase. J. Neurosci. 17, 7462–7470 (1997).
Wong, T. H. et al. PRKAR1B mutation associated with a new neurodegenerative disorder with unique pathology. Brain. 137, 1361–1373 (2014).
Pottier, C., Baker, M., Dickson, D. W. & Rademakers, R. PRKAR1B mutations are a rare cause of FUS negative neuronal intermediate filament inclusion disease. Brain. 138, e357 (2015).
Geisheker, M. De novo missense mutations in neurodevelopmental disorders. ResearchWorks Archive. University of Washington. https://digital.lib.washington.edu/researchworks/handle/1773/44273 (2019).
Hormozdiari, F., Penn, O., Borenstein, E. & Eichler, E. E. The discovery of integrated gene networks for autism and related disorders. Genome Res. 25, 142–154 (2015).
Coe, B. P. et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat. Genet. 51, 106–116 (2019).
Kaplanis, J. et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 586, 757–762 (2020).
Ruzzo, E. K. et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 178, 850–66 e826 (2019).
Espiard, S. et al. Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight. 3, e98296 (2018).
Schernthaner-Reiter, M. H., Trivellin, G. & Stratakis, C. A. Interaction of AIP with protein kinase A (cAMP-dependent protein kinase). Hum. Mol. Genet. 27, 2604–2613 (2018).
Drougat, L. et al. Genomic and sequence variants of protein kinase A regulatory subunit type 1beta (PRKAR1B) in patients with adrenocortical disease and Cushing syndrome. Genet. Med. 23, 174–182 (2021).
Nesterova, M., Yokozaki, H., McDuffie, E. & Cho-Chung, Y. S. Overexpression of RII beta regulatory subunit of protein kinase A in human colon carcinoma cell induces growth arrest and phenotypic changes that are abolished by site-directed mutation of RII beta. Eur. J. Biochem. 235, 486–494 (1996).
Mavrakis, M., Lippincott-Schwartz, J., Stratakis, C. A. & Bossis, I. Depletion of type IA regulatory subunit (RIalpha) of protein kinase A (PKA) in mammalian cells and tissues activates mTOR and causes autophagic deficiency. Hum. Mol. Genet. 15, 2962–2971 (2006).
Rada-Iglesias, A., Bajpai, R., Prescott, S., Brugmann, S. A., Swigut, T. & Wysocka, J. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell. 11, 633–648 (2012).
Zhang, K. et al. Imbalance of excitatory/inhibitory neuron differentiation in neurodevelopmental disorders with an NR2F1 point mutation. Cell. Rep. 31, 107521 (2020).
Laugsch, M. et al. Modeling the pathological long-range regulatory effects of human structural variation with patient-specific hiPSCs. Cell Stem Cell. 24, 736–52.e712 (2019).
Ilouz, R. et al. Localization and quaternary structure of the PKA RIbeta holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 109, 12443–12448 (2012).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581, 434–443 (2020).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Katsonis, P. & Lichtarge, O. A formal perturbation equation between genotype and phenotype determines the evolutionary action of protein-coding variations on fitness. Genome Res. 24, 2050–2058 (2014).
Levenson, J. M., O’Riordan, K. J., Brown, K. D., Trinh, M. A., Molfese, D. L. & Sweatt, J. D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40545–40559 (2004).
Acknowledgements
Research reported in this paper was partially supported by the National Institutes of Health (NIH) Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under award number U01HG007703 (SFN, JAM-A, CGSP) and the UCLA California Center for Rare Diseases, within the Institute of Precision Health. Part of this work was supported by the intramural research program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), NIH through a grant to C.A.S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The paper includes important contributions from On behalf of the Members of the Undiagnosed Diseases Network. The human embryonic material was provided by the Joint MRC/Wellcome (MR/R006237/1) Human Developmental Biology Resource.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization: F.M., G.S., C.A.S., C.P.S. Data curation: F.E., A. Sievers, P.K., O.L. Formal analysis: F.E., P.K., A. Sievers, E.L., N.S., C.A.S. Funding acquisition: C.P.S., O.L., C.A.S. Investigation: F.M., C.P.S., G.S., C.A.S., N.S., E.L., J.A.R., E.T., C.H., E.S., E.K., S.C., S.F.N., J.A.M., C.G.S.P., R.H.S., M.V.A., D.K.G., R.W., R.P., A.T., M.L., S.T., Y.C., O.L., P.K., A. Stocco. Methodology: F.M., C.P.S., P.K., C.A.S., Y.C. Project administration: F.M., C.P.S. Resources: Human Developmental Biology Resource; Software: O.L., P.K. Supervision: F.M., C.P.S. Visualization: F.M., P.K., C.A.S., E.L., N.S., Y.C. Writing—original draft: F.M., G.S., C.P.S. Writing—review & editing: F.M., C.P.S., G.S., F.E., C.A.S., N.S., E.L., J.A.R., E.T., C.H., E.S., E.K., S.C., S.F.N., J.A.M., C.G.S.P., R.H.S., M.V.A., D.K.G., R.W., R.P., A.T., M.L., S.T., Y.C., O.L., P.K., A. Stocco.
Corresponding author
Ethics declarations
Ethics declaration
Data collection was performed under the umbrella of human research study H-34578 (Understanding the Molecular Causes of Neuropsychiatric Disease), which was approved by the Baylor College of Medicine Institutional Review Board (IRB). Written informed consent for publication of medical data and images was obtained from the parents or legal guardians of the respective individuals as required by the IRB.
Competing interests
E.T., R.W., R.P., and A.T. are employees of GeneDx, Inc. J.A.R., O.L., and P.K. are employees of Baylor College of Medicine. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from clinical genetic testing conducted at Baylor Genetics Laboratory. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marbach, F., Stoyanov, G., Erger, F. et al. Variants in PRKAR1B cause a neurodevelopmental disorder with autism spectrum disorder, apraxia, and insensitivity to pain. Genet Med 23, 1465–1473 (2021). https://doi.org/10.1038/s41436-021-01152-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01152-7
This article is cited by
-
Protein Kinase A in neurological disorders
Journal of Neurodevelopmental Disorders (2024)
-
High-dimensional phenotyping to define the genetic basis of cellular morphology
Nature Communications (2024)
-
Genetic pain loss disorders
Nature Reviews Disease Primers (2022)
-
Alpha-Synuclein, cyclooxygenase-2 and prostaglandins-EP2 receptors as neuroinflammatory biomarkers of autism spectrum disorders: Use of combined ROC curves to increase their diagnostic values
Lipids in Health and Disease (2021)
