
Abstract
Purpose
Genetic testing for pediatric cancer predisposition syndromes (CPS) could augment newborn screening programs, but with uncertain benefits and costs.
Methods
We developed a simulation model to evaluate universal screening for a CPS panel. Cohorts of US newborns were simulated under universal screening versus usual care. Using data from clinical studies, ClinVar, and gnomAD, the presence of pathogenic/likely pathogenic (P/LP) variants in RET, RB1, TP53, DICER1, SUFU, PTCH1, SMARCB1, WT1, APC, ALK, and PHOX2B were assigned at birth. Newborns with identified variants underwent guideline surveillance. Survival benefit was modeled via reductions in advanced disease, cancer deaths, and treatment-related late mortality, assuming 100% adherence.
Results
Among 3.7 million newborns, under usual care, 1,803 developed a CPS malignancy before age 20. With universal screening, 13.3% were identified at birth as at-risk due to P/LP variant detection and underwent surveillance, resulting in a 53.5% decrease in cancer deaths in P/LP heterozygotes and a 7.8% decrease among the entire cohort before age 20. Given a test cost of $55, universal screening cost $244,860 per life-year gained; with a $20 test, the cost fell to $99,430 per life-year gained.
Conclusion
Population-based genetic testing of newborns may reduce mortality associated with pediatric cancers and could be cost-effective as sequencing costs decline.
Similar content being viewed by others

INTRODUCTION
Universal newborn screening (NBS) has successfully decreased the morbidity and mortality of a wide range of severe pediatric-onset diseases including phenylketonuria, cystic fibrosis, and sickle cell disease.1 Genetic testing has the potential to augment universal NBS programs, and research exploring the medical, technological, public health, and ethical implications of universal newborn genetic screening is ongoing.2,3 Detection of germline pathogenic variants in genes associated with a high risk of early childhood tumors could be incorporated into expanded NBS programs; variant detection would prompt application of accepted clinical care recommendations currently utilized by pediatric oncologists for infants and children with known cancer predisposition syndromes (CPS).4
Decision modeling can evaluate the potential of genetic testing in NBS, as it can facilitate evidence synthesis, provide data to inform clinical guidelines,5,6 and evaluate new diseases for inclusion in NBS.7 This is especially useful in settings of rare diseases, like pediatric cancer, where sufficiently powered randomized clinical trials that test the clinical utility of NBS for early onset disease would be difficult. Using a decision-analytic framework, we asked: what are the potential clinical benefits, harms, and cost-effectiveness of newborn genetic screening, using a targeted next-generation sequencing (t-NGS) approach for a select panel of genes associated with early onset childhood cancer?
MATERIALS AND METHODS
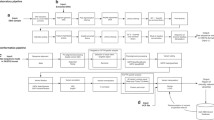
We developed the Precision Medicine Policy and Treatment (PreEMPT) model to estimate the potential risks and benefits of population-based genetic screening for pathogenic germline variants in RET, RB1, TP53, DICER1, SUFU, PTCH1, SMARCB1, PHOX2B, ALK, WT1, or APC. These autosomal dominant cancer predisposition genes were selected because of their association with very early onset malignancy and the availability of surveillance guidelines for early detection starting in infancy8,9,10,11,12,13,14 (Table 1). Using data from clinical studies, ClinVar,15 the Genome Aggregation Database (gnomAD),16 and the US Surveillance, Epidemiology and End Results (SEER) Program,17 we assigned each newborn a probability of carrying a pathogenic or likely pathogenic (P/LP) variant in each of the 11 genes (RET variants were restricted to those for multiple endocrine neoplasia type 2B [MEN2B]18). We limited variants to high-quality P/LP variants identified in ClinVar (i.e., 2-star) and confirmed the list via curation by the Mass General Brigham Laboratory for Molecular Medicine (Supplemental Tables 1, 2). The prevalence of P/LP variants was based on best available data from clinical studies for cancer cases and gnomAD data for noncancer cases (Supplemental Table 3). For genes without any P/LP variants in gnomAD, we assumed an allele frequency of 0.5 among the 282,912 alleles in gnomAD. We assumed that the occurrence of P/LP variants were independent and summed allele frequencies across all variants for each gene. Using Bayes’ theorem to synthesize data on the proportions of individuals with and without P/LP variants and who develop each cancer before age 20, we estimated the penetrance for each gene, defined as the probability an individual carrying a P/LP variant will develop a condition before age 20. See Supplemental Materials for additional details.
Cohorts of newborns representative of a modern US birth cohort were simulated under the scenarios of usual care and t-NGS at birth and followed throughout their lifetimes (Supplemental Fig. 1). Under t-NGS, we assumed newborns with identified P/LP variants would undergo cancer surveillance based on established guidelines (Table 1). As a best-case estimate of program efficacy, we assumed 100% adherence with t-NGS screening and surveillance recommendations.
Utilizing SEER data and published literature to estimate incidence, stage distribution, and outcomes, newborns were at risk for each cancer of interest (those associated with the 11 CPS). Treatment by stage for each diagnosis was based on standard care and included radiation and chemotherapy when indicated (Supplemental Table 4). Individuals who received chemotherapy and/or radiation as part of cancer treatment faced excess late mortality risks as adults starting at age 20 based on the Childhood Cancer Survivor Study.19,20 Newborns found to be heterozygotes of P/LP variants underwent surveillance, which resulted in early detection of malignancy, which, for specific cancers, resulted in reduced use of radiation and/or chemotherapy and improved outcomes (Table 1). Clinical benefit for t-NGS and associated surveillance was modeled as reductions in advanced disease, cancer deaths, and treatment-related late mortality.
Costs were estimated for direct medical costs, patient time costs, and genetic testing (for the t-NGS strategy) (Supplemental Table 5). For t-NGS, we assumed a cost of $55 for the 11-gene panel test (i.e., $5 per gene) based upon expert input, current cost of NBS, and commercial cost for a panel test.21 We assumed that this cost reflected the incremental cost of adding the panel to a NBS program with existing infrastructure for genetic testing. Costs for surveillance and cancer treatment were based on published estimates and national databases. To account for patient time costs, we included parental time lost from work (see Supplemental Materials). All costs were expressed in 2018 dollars.
To capture uncertainty, we conducted 1,000 simulations in which each parameter was sampled from its underlying distribution and report the mean and 95% uncertainty interval (UI) for all outcomes. Given the goals of improving child health, the model did not include impact of early detection of adult-onset malignancy, which is increased in some CPS (for example, TP53), or impact on family member health/reflex genetic testing.
To assess the potential harms associated with t-NGS, most prominently the burden of a genetic diagnosis in the absence of a pediatric cancer occurrence, we defined individuals with P/LP variants who developed cancer before age 20 (true positives) as having “penetrant variant status” (PVS) and individuals with P/LP variants who did not develop cancer by age 20 as having “nonpenetrant variant status” (NPVS). This allowed us to illustrate the harm–benefit tradeoffs associated with t-NGS by estimating the number of NPVSs per PVS, cancer death averted, and life-year (LY) gained in the cohort.
To assess the cost-effectiveness of genetic CPS screening, we calculated an incremental cost-effectiveness ratio (ICER), defined as the additional cost of t-NGS divided by its additional clinical benefit compared with usual care, expressed as cost per LY gained. Although higher cost-effectiveness thresholds have been suggested for rare diseases,22 we estimated the threshold cost for the 11-gene panel test at which t-NGS would achieve an ICER of <$100,000 per LY gained as changes in technology will likely impact costs.
Sensitivity analyses examined the influence of assumptions on P/LP variant prevalence among cancer and noncancer cases, adherence to guideline surveillance, and surveillance and cancer treatment costs, as well as stage-specific estimates of 5-year survival, the proportion of cancers diagnosed as advanced disease, and excess mortality risks associated with cancer treatment.
RESULTS
In a typical US birth cohort of 3.7 million newborns, the model estimated 1,803 individuals would develop a CPS-associated cancer before age 20 (95% UI, 1,756 to 1,850), 13.3% of whom would have P/LP CPS variants (95% UI, 11.3% to 15.7%). Under t-NGS, 1,584 individuals with P/LP CPS variants (95% UI, 1,230 to 2,026) would be identified, 232 (95% UI, 196 to 278) of whom would develop cancer before age 20 (i.e., PVS) and 1,353 (95% UI, 991 to 1,788) would not (i.e., NPVS). This resulted in an estimated positive predictive probability, or penetrance, of 14.8% (95% UI, 11.2% to 19.6%) for the 11-gene panel and a relative risk of developing a cancer before the age of 20 of 351 (95% UI, 260 to 468) among individuals with P/LP variants. Penetrance and relative risk estimates varied for individual genes (Table 2). In terms of clinical benefit, the model estimated that compared with usual care, t-NGS would reduce cancer deaths before age 20 overall by 7.8% (95% UI, 5.8% to 10.1%) and decrease the proportion of 5-year survivors at risk for radiation-related excess mortality by 5.8% (95% UI, 3.6% to 8.6%) (Table 2). Additionally, t-NGS would increase the number of adult cancer survivors alive at age 45 by 2.1% (95% UI, 1.4 to 2.9%), and result in a gain of 2,937 (95% UI, 2,244 to 3,879) LY. The estimated benefit for all outcomes was considerably higher among individuals with P/LP variants (Fig. 1). For example, among P/LP heterozygotes, t-NGS would reduce cancer deaths before age 20 by 53.5% (95% UI, 47.1% to 60.5%). In terms of harm–benefit tradeoffs, for t-NGS, the number with NPVS identified per PVS was 5.9 (95% UI, 4.1 to 7.9), the number of NPVS per cancer death averted was 43.5 (95% UI, 29.1 to 61.5), and the number of NPVS per LY gained was 0.5 (95% UI, 0.3 to 0.7). Compared with usual care, t-NGS had an ICER of $244,860 per LY gained (95% UI, $181,500/LY to $327,520/LY) assuming a 11-gene panel cost of $55 per newborn. At a panel cost of $20, the ICER fell to $99,430 per LY (95% UI, $72,510/LY to $137,330/LY).

Shown are estimates for the overall cohort of 3.7 million screened newborns and among the subset of newborns with identified pathogenic/likely pathogenic (P/LP variants). The 95% uncertainty intervals among the 1,000 simulations are listed in the parantheses and shown by the bars.
Cost-effectiveness of t-NGS was most sensitive to P/LP variant prevalence among cancer cases and differences in 5-year survival rates for localized versus advanced disease, moderately sensitive to the proportion of cancers diagnosed with advanced disease and the P/LP variant prevalence among controls, and robust to assumptions on surveillance and cancer treatment costs (Supplemental Fig. 2). With less than full adherence to surveillance guidelines, the ICER for t-NGS increased to $270,260/LY with 90% compliance (95% UI, $201,160/LY to $361,210/LY) and $321,000/LY with 70% compliance (95% UI, $240,480/LY to $428,720/LY).
DISCUSSION
Leveraging data from ClinVar, gnomAD, SEER, and published literature, we used a model-based approach to estimate the potential clinical impact of universal genetic screening in newborns for pediatric CPS. Our findings suggest that under the best-case assumption of full adherence to screening and surveillance guidelines, t-NGS would identify approximately 1,580 individuals with P/LP CPS variants among 3.7 million newborns each year in the United States. If these newborns were evaluated, underwent genetic counseling, and offered cancer surveillance, more than half of cancer deaths among individuals with CPS variants would be averted. Further, as the costs of genetic screening decline, targeted newborn screening for pediatric cancer genes could be cost-effective given benchmarks for “good value.”23
Newborn screening for any disorder requires balancing the potential benefits (prevention or early detection of disease) and harms (unnecessary surveillance costs and parental anxiety). Inclusion of genetic testing for CPS risk as part of NBS programs will present new uncertainties, most importantly with respect to the “allowable” burden of tests detecting P/LP variants of unknown or low penetrance (e.g., parents who are told that the infant is at increased risk of cancer, but cancer may not manifest in childhood or at all). While we modeled 11 CPS genes as a panel, analyses on individual or subsets of genes can guide efforts to reduce potential harm by identifying genes with higher penetrance (e.g., RB1) or where the benefit is well understood (RET). Of importance, in our study, we assumed that the CPS test would be included as part of state-wide NBS programs, after completion of successful pilot testing. We recognize, however, that the process of adding new tests is complex and varies by state. In an alternative model, separate consent for this test (outside of usual NBS) would create added burden and require additional resources for implementation not reflected in our study.
While we provided estimates of the potential harm–benefit tradeoffs, we did not account for the impact of this knowledge on families, as well as other potential impacts of testing, such as risk of adult-onset cancer, family reproductive planning, and detection of cancer risk in family members; future studies should consider these important factors. Available data from families with CPS suggest that entering a child into a surveillance protocol based upon genetic testing decreased anxiety and did not create an excessive burden.24
We assumed full adherence to surveillance recommendations to estimate the potential survival benefit of surveillance for CPS-associated pediatric cancers. As the cost-effectiveness of screening was less favorable with lower adherence, ensuring adherence will be crucial to realize the projected benefits. Of note, other benefits of early detection to avert toxicity (such as avoidance of blindness after early detection of retinoblastoma) were not captured in our model.
We assumed, in our cost estimates, that NBS will move forward nationally to establish infrastructure that supports genetic screening in general. Additional resources will be needed to build this capacity, as well as support for families after genetic information disclosure. The benefits of surveillance, as modeled in our study, are based upon scant data, but represent current recommendations for clinically detected children with CPS. The National Cancer Institute (NCI) Childhood Cancer Data Initiative aims to collect data on every child diagnosed with cancer in the United States and may provide more precise data in the coming years. Our model can readily incorporate these and other new data as they become available to generate updated estimates.
While our findings are suggestive, using newborn genetic screening for pediatric CPS as an example, our study demonstrates how advances in genetics can be applied to populations, and what the implications might be for public health. Prospective clinical studies that investigate crucial factors—such as parental uptake of testing, impact of a genetic CPS diagnosis on families, adherence to surveillance, and effectiveness of surveillance in preventing advanced disease—are necessary before this testing can be proposed as a component of population-based newborn screening.
Data availability
Additional details about the Precision Medicine Policy and Treatment (PreEMPT) model, data used as model input parameters, and output data from the model are available by request from the corresponding author. A list of variants included in the modeling study is also provided in Supplemental Table 2.
References
Centers for Disease Control and Prevention. Impact of expanded newborn screening—United States, 2006. MMWR Morb. Mortal. Wkly Rep. 57, 1012–1015 (2008).
Holm, I. A. et al. The BabySeq project: implementing genomic sequencing in newborns. BMC Pediatr. 18, 225 (2018).
Berg, J. S. et al. Newborn sequencing in genomic medicine and public health. Pediatrics 139, e20162252 (2017).
Brodeur, G. M., Nichols, K. E., Plon, S. E., Schiffman, J. D. & Malkin, D. Pediatric cancer predisposition and surveillance: an overview, and a tribute to Alfred G. Knudson Jr. Clin. Cancer Res. 23, e1–e5 (2017).
Owens, D. K. et al. Use of decision models in the development of evidence-based clinical preventive services recommendations: methods of the U.S. Preventive Services Task Force. Ann. Intern. Med. 165, 501–508 (2016).
Payne, K., Gavan, S. P., Wright, S. J. & Thompson, A. J. Cost-effectiveness analyses of genetic and genomic diagnostic tests. Nat. Rev. Genet. 19, 235–246 (2018).
Prosser, L. A., Grosse, S. D., Kemper, A. R., Tarini, B. A. & Perrin, J. M. Decision analysis, economic evaluation, and newborn screening: challenges and opportunities. Genet. Med. 14, 703–712 (2013).
Wasserman, J. D. et al. Multiple endocrine neoplasia and hyperparathyroid-jaw tumor syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin. Cancer Res. 23, e123–e132 (2017).
Kamihara, J. et al. Retinoblastoma and neuroblastoma predisposition and surveillance. Clin. Cancer Res. 23, e98–e106 (2017).
Kratz, C. P. et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin. Cancer Res. 23, e38–e45 (2017).
Foulkes, W. D. et al. Cancer surveillance in Gorlin syndrome and rhabdoid tumor predisposition syndrome. Clin. Cancer Res. 23, e62–e67 (2017).
Schultz, K. A. P. et al. DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin. Cancer Res. 24, 2251–2261 (2018).
Kalish, J. M. et al. Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin. Cancer Res. 23, e115–e122 (2017).
Achatz, M. I. et al. Cancer screening recommendations and clinical management of inherited gastrointestinal cancer syndromes in childhood. Clin. Cancer Res. 23, e107–e114 (2017).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
National Cancer Institute. Surveillance, Epidemiology and End Results (SEER) Program. SEER*Stat Database. Incidence-based mortality SEER 18 regs (excl Louisiana) research data, Nov 2016 sub (2000-2014) —linked to county attributes—total U.S., 1969-2016 counties. https://seer.cancer.gov/datasoftware/documentation/seerstat/nov2016/ (2016).
Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Pediatrics 142, e20183062 (2018).
Yeh, J. M. et al. Life expectancy of adult survivors of childhood cancer over 3 decades. JAMA Oncol. 6, 350–357 (2020).
Armstrong, G. T. et al. Reduction in late mortality among 5-year survivors of childhood cancer. N. Engl. J. Med. 374, 833–842 (2016).
Invitae. Invitae pediatric solid tumors panel. https://www.invitae.com/en/physician/tests/01104/ (2020).
Prosser, L. A. Defining the value of treatments of rare pediatric conditions. JAMA Pediatr. 172, 1123–1124 (2018).
Neumann, P. J., Cohen, J. T. & Weinstein, M. C. Updating cost-effectiveness-the curious resilience of the $50,000-per-QALY threshold. N. Engl. J. Med. 371, 796–797 (2014).
Duffy, K. A., Grand, K. L., Zelley, K. & Kalish, J. M. Tumor screening in Beckwith-Wiedemann syndrome: parental perspectives. J. Genet. Couns. 27, 844–853 (2018).
Acknowledgements
We thank Matt Lebo, Sami Amr, Lorena Lazo de la Vega, Lisa Marie Mahanta, and Natascia Anastasio for curating the variant list.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.M.Y., N.K.S., K.D.C., P.M.M., C.L.B.Z., R.C.G., C.Y.L., H.L.R., M.S.W., L.D., A.C.W. Data curation: J.M.Y., N.K.S., A.C., K.D.C., P.M.M., M.G., C.L.B.Z., L.D., A.C.W. Formal analysis: J.M.Y., N.K.S., K.D.C., P.M.M., G.O., M.G., N.R., L.D., A.C.W. Funding acquisition: J.M.Y., N.K.S., R.C.G., A.C.W. Investigation: J.M.Y., N.K.S., A.C., K.D.C., P.M.M., G.O., N.R., C.L.B.Z., R.C.G., H.L.R., M.S.W., L.D., A.C.W. Methodology: J.M.Y., N.K.S., K.D.C., P.M.M., M.G., A.C.W. Visualization: J.Y., G.O., N.R. Writing—original draft: J.M.Y., N.K.S., K.D.C., P.M.M., G.O., C.L.B.Z., R.C.G., H.L.R., M.S.W., L.D., A.C.W. Writing—review & editing: J.M.Y., N.K.S., A.C., K.D.C., M.G., P.M.M., G.O., N.R., C.L.B.Z., R.C.G., C.Y.L., H.L.R., M.S.W., L.D., A.C.W.
Corresponding author
Ethics declarations
Competing interests
This project was funded by the National Institutes of Health (NIH) (5R01HD090019–04, principal investigator [PI] A.C.W.), which had no role in the design of the study, collection and analysis of data and decision to publish. K.D.C. was supported by NIH grant K01-HG009173. R.C.G. is co-founder of Genome Medical, Inc. and has received compensation for advising AIA, Grail, Humanity, Kneed Media, Plumcare, UnitedHealth, Verily, VibrentHealth, and Wamberg. H.L.R. serves on the Scientific Advisory Board for Genome Medical, Inc. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yeh, J.M., Stout, N.K., Chaudhry, A. et al. Universal newborn genetic screening for pediatric cancer predisposition syndromes: model-based insights. Genet Med 23, 1366–1371 (2021). https://doi.org/10.1038/s41436-021-01124-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01124-x
