
Abstract
Purpose
ClinGen provides gene-specific guidance for interpretation of sequence variants in MYH7. We assessed laboratory and clinical impact of reclassification by the American College of Medical Genetics and Genomics-Association for Molecular Pathology (ACMG-AMP) and ClinGen recommendations in 43 MYH7 variants reported by a diagnostic laboratory between 2013 and 2017.
Methods
Fifty-two proband reports containing MYH7 variants were reinterpreted by original ACMG-AMP and ClinGen guidelines. Evidence items were compared across schemes and reasons for classification differences recorded. Laboratory impact was assessed by number of recommended report reissues, and reclassifications coded as clinically “actionable” or “equivalent.” Available pedigrees were reviewed to describe projected cascade impact.
Results
ClinGen produced a higher proportion of diagnostic classifications (65% of variants) compared with ACMG-AMP (54%) and fewer variants of uncertain significance (30% versus 42%). ClinGen classification resulted in actionable changes in 18% of variants with equal upgrades and downgrades from original report. ClinGen’s revisions to PM1 and PS4 contributed to classification differences in 21% and 19% of variants respectively. Each classification change per proband report impacted, on average, 3.1 cascade reports with a further 6.3 first- and second-degree relatives potentially available for genotyping per family.
Conclusion
ClinGen’s gene-specific criteria provide expert-informed guidance for interpretation of MYH7 sequence variants. Periodic re-evaluation improves diagnostic confidence and should be considered by clinical and laboratory teams.
Similar content being viewed by others

INTRODUCTION
Consistent and accurate interpretation of sequence variants is essential for effective clinical care for individuals with inherited cardiomyopathy and their families. Hypertrophic cardiomyopathy (HCM) is common and demonstrates considerable clinical and genetic heterogeneity1,2, with presentations ranging from mild or asymptomatic disease to sudden cardiac death in the young. Pathogenic (P) or likely pathogenic (LP) variants in MYH7 are identified in approximately a third of individuals with HCM3,4, with rare or novel variants accounting for almost half of these5. Accurate and timely molecular characterization permits diagnostic security and may guide risk stratification and management6.
Widespread utilization of next-generation sequencing (NGS) in HCM has uncovered substantial genetic variation and highlighted the importance of a standardized approach to variant interpretation. Determination of pathogenicity is a complex process involving distillation of various pieces of information from multiple sources that may change over time and are subject to variable interpretation and application. Laboratories and medical geneticists must be adaptive and implement strategies to stay abreast of new information as it becomes available and modify reporting practices to ensure clinicians and patients have up-to-date and clinically relevant information.
Revised consensus recommendations by the American College of Medical Genetics and Genomics-Association for Molecular Pathology (ACMG-AMP)7 provide a framework for clinical interpretation of sequence variants that is now widely utilized8. Prior to implementation of these guidelines laboratories frequently employed bespoke or ad hoc classification practices that evolved over time and were inconsistent between laboratories. Additionally, availability of robust population data (Exome Aggregation Consortium9 and Genome Aggregation Database)10,11 has permitted improved discrimination between disease-causing variants and benign variation.
The 2015 ACMG-AMP guidelines were designed to be widely applicable, but several elements lack specificity12,13, are applied discordantly,14,15 and do not take into account disease-specific mechanisms. The Clinical Genome Resource (ClinGen)16 aims to leverage expert knowledge of genetic variation and molecular underpinnings in specific disease groups to develop guidelines that take into account gene-specific information. In 2017, ClinGen’s Inherited Cardiomyopathy Expert Panel published modified guidelines for interpretation of sequence variants in MYH7-associated HCM17. These recommendations provided increased specificity, clarified interpretation of particular ACMG-AMP evidence items, and resulted in expert curation and submission of 60 MYH7 variants to the ClinVar database18. The pilot recommendations also provided impetus for similar guidelines in other disorders19,20,21 (including recent refinement for dilated cardiomyopathy22) and further modification using phenotype-enhanced criteria23. There has been limited independent evaluation of the ClinGen MYH7-specific guidelines, although a recent study demonstrated a nonsignificant trend toward decreased variant of uncertain significance (VUS) burden in two HCM cohorts23.
Clinical reporting of variants is probabilistic; the inherent uncertainty in test results has clinical and psychoemotional implications for the individual and the treating clinician. In particular, VUS have limited clinical utility and may result in confusion and anxiety for individuals and families24,25. Equally, misattribution of causation has the potential to impact negatively on clinical care or result in unnecessary investigations for unaffected family members. Confidence in genetic test results is influenced by recency of reporting and information available at the time of the report. Consequently, a gradient of confidence exists that brings into question many variants classified prior to endorsement of the ACMG-AMP guidelines and availability of population databases. Resultant uncertainty in assignment of pathogenicity in older reports furthermore complicates interpretation of current test results, which necessarily take into account previous classifications. Periodic review of previously classified variants with availability of new information and contemporary guidelines may improve diagnostic confidence and provides a mechanism for clarification of VUS3,26,27. Such review also permits iterative refinement of curation processes and enrichment of local and public clinical databases with up-to-date evidence and classifications.
This study evaluates the projected clinical and reporting impact of reclassification of MYH7 variants in a cohort of individuals with cardiomyopathy, arrhythmia, and/or sudden death ascertained through diagnostic genetic testing undertaken in a single clinical laboratory. MYH7 variants were re-evaluated by both unmodified ACMG-AMP criteria (using information currently available) and ClinGen’s modified MYH7-specific guidelines, providing a basis for comparison of these two schemes and interrogation of the items most commonly responsible for changes in classification.
MATERIALS AND METHODS
Data collection
Proband reports issued through a clinically accredited (by the National Association of Testing Authorities [NATA]) laboratory in Victoria, Australia, between 2013 and 2017 were queried for reports of at least one MYH7 variant (54 variants across 52 proband reports). These reports represented individuals who had undergone diagnostic testing using gene panels for cardiomyopathy, arrhythmia, and/or sudden death. Reports issued for cascade and predictive reasons were excluded. During this period, the laboratory performed testing by custom capture using Agilent Sure Select (Agilent Technologies, Santa Clara, CA, USA) followed by sequencing on an Illumina machine (MiSeq or HiSeq; Illumina, San Diego, CA, USA). Variant reports issued prior to availability of the 2015 ACMG-AMP criteria7 were typically classified according to the body of evidence available at the time and in consideration of the interpretive categories defined in the earlier and less prescriptive ACMG recommendations28. Formal adoption of the ACMG-AMP guidelines occurred in early 2016. The query identified 52 proband reports that met criteria. These were reviewed and brief demographic details, indication for testing, genotype, and reported variant classification was recorded using a custom REDCap electronic data capture tool29 hosted at Murdoch Children’s Research Institute (Melbourne, Australia). The project received Human Research Ethics Committee approval (HREC/52933/RCHM-2019).
Curation and classification
All MYH7 variants were curated and classified according to original unmodified ACMG-AMP criteria (“original ACMG”) and ClinGen’s MYH7-specific guidelines (“ClinGen”). The original ACMG-AMP scheme was applied strictly and where feasible was not informed by evidence arising primarily through the ClinGen paper17. For example, moderate (domain) evidence was rarely selected per the original ACMG-AMP scheme, as ClinGen introduced the ability to select moderate evidence (PM1) for missense variants occurring in the 181–937 amino acid residue hotspot5,30. Similarly, the original scheme does not define number of affected probands required for strong clinical evidence (PS4) selection, so this was applied only where specific wording of the ACMG-AMP criteria was met. ClinGen-modified PS4 criteria requiring “probands with consistent phenotype” were interpreted to mean unrelated individuals with HCM, as reported in published medical literature, clinical databases (ClinVar18), or on review of the medical record, where accessible, by the curating physician. For variants previously classified by ClinGen, the curator was blinded to the ClinGen classification at the outset of curation, although this was typically uncovered as part of literature review and curation. Effort was made to apply ClinGen criteria agnostic of the previously assigned classification. ClinGen criteria were applied according to the original MYH7-specific guidelines17, and did not incorporate revisions recommended subsequent to completion of curation (for example, proposed reduced weighting of absence/rarity PM2 criterion31).
For both original ACMG-AMP and ClinGen schemes, items of evidence selected were coded and final classification recorded. Classifications by each scheme were compared with each other and with the initial (“report”) classification. Where differences occurred, the reason for the classification change was coded and responsible evidence items annotated. Variants in which a classification change occurred (“report” and “ClinGen” classifications differed) were discussed in a multidisciplinary team meeting attended by genetic scientists, clinical geneticists, and genetic counselors to achieve consensus regarding final classification. Curation occurred between March 2019 and March 2020.
Laboratory impact was assessed on the basis of whether a report reissue was recommended (“no report change” or “report change”), and number of family members previously cascade tested was recorded. A report reissue was recommended in all cases where ClinGen and report classifications were discordant, and the direction of the change was coded. Classification changes were defined as clinically actionable if they occurred between VUS and LP/P categories, or between LP/P and benign (B) or likely benign (LB) categories. Classification differences between LP and P categories were considered clinically equivalent. Where classification changes occurred, clinical files were reviewed (as available; n = 9) to assess number of family members genotyped through the laboratory and expected clinical impact. Pedigrees were reviewed (where available; n = 6) to determine the number of untested first- and second-degree relatives available for cascade testing. Values were reported as mean number of individuals per family, with associated range. Descriptive statistics were generated using SPSS Statistics v26 (IBM, NY, USA).
RESULTS
Descriptive data
MYH7 variants were reported 54 times across 52 separate proband reports (with two instances of a report listing two MYH7 variants in the same individual). These represented 43 unique variants, 37 (86%) of which were reported only once and 6 (14%) were recurrent within the call set. All reported variants were single-nucleotide variants. Forty (93%) of these were missense and 3 (7%) were splice site variants, consistent with the reported mutational spectrum of MYH7. Segregation information was not available for 74% of variants but 10 (19%) were inherited and 4 (7%) were apparently de novo (paternity assumed). The average number of variants listed per report, including those in other genes, was 2.1 (range 1–6).
In seven cases (14%) an amended report had previously been issued with an updated variant classification (for example, due to segregation or other new information). Where this occurred, both initial and most recent classifications were captured and the most recent classification used in all comparisons. One locally recurrent variant was classified differently on two separate reports creating an additional data point for comparisons between original report classifications and both ACMG-AMP/ClinGen classifications (n = 44). Thirteen (30%) of the unique variants have previously been curated by ClinGen17.
Classification description and distribution
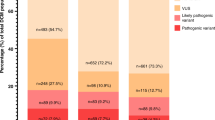
Application of ClinGen criteria resulted in a higher number of diagnostic classifications (LP or P) compared with original ACMG-AMP. The original ACMG-AMP scheme resulted in diagnostic classifications in 56% of reports and 54% of variants. Both initial report and the ClinGen scheme demonstrated LP/P outcomes in 63% of reports (65% of variants) although there were discrepant classifications in both directions. Classifications of pathogenicity by initial report, ACMG-AMP, and ClinGen schemes are summarized in Table 1, organized by reported variants (n = 54) and by unique variants (n = 43). A complete list of variants classified, and revised ClinGen criteria selected, is presented in Supplementary Data 1. The frequency with which individual evidence items were used to inform final classification is presented in Table 2 for both schemes.
Original report comparisons
Classification by strict ACMG-AMP criteria resulted in classification changes in 23 reports (43%) and 17 variants (39%). Ten report (19%) and variant (23%) classification differences represented clinically actionable changes. There were three instances (6% of reports and 7% of variants) where a new molecular diagnosis was made (VUS upgraded to LP/P) and seven cases (13% of reports and 16% of variants) where a molecular diagnosis was revoked (LP/P to VUS or benign). Concordance in classification between original report and ACMG-AMP/ClinGen schemes is summarized in Fig. 1.

Comparison of (a) original report versus American College of Medical Genetics and Genomics (ACMG-AMP) classification, (b) original report versus ClinGen classification, and (c) ACMG-AMP versus ClinGen classification, organized by number of proband reports (left grid) and by variants (right grid). Bubble plots denote the respective comparisons with area of the bubble proportional to the number of variants. B benign, LB likely benign, LP likely pathogenic, P pathogenic, VUS variant of uncertain significance. aRepresents 43 unique variants; however, one locally recurrent variant was classified differently on two separate initial reports, necessitating an additional comparison point (n = 44).
Several factors contributed to differences in classification (Table 3). Classification changes in eight variants (47%) with ACMG-AMP criteria applied occurred due to availability of new information since original report issue (for example population data, clinical reports, segregation). Differences that reflected scheme application only, or differences in evidence weighting or scoring matrices that were independent of availability of new information, were considered “scheme-specific differences.” These were a factor in classification changes for nine variants (53%) by ACMG-AMP criteria, of which eight were reported in 2015 or earlier, prior to formal implementation of the ACMG-AMP criteria locally.
Application of ClinGen criteria resulted in a classification change from 19 original reports (35%) of which eight changes (15%) were clinically actionable. These changes represented 13 variants (30%), 8 of which (18%) were clinically actionable changes. Of the actionable changes, a new molecular diagnosis was achieved in half (7% of probands, 9% of variants) and molecular diagnosis revoked in half. Thirteen variants included in this study were also classified in the paper that described the ClinGen scheme17; in all cases the classifications were concordant. Classification differences between the original report and the ClinGen scheme reflected specific elements of the scheme for seven variants (54%) and were attributable to new information emerging with passage of time in six variants (46%). There was a trend toward older reports being more likely to undergo a change in classification, although this was not statistically significant (χ2 1.7, p > 0.5). Forty-four percent of reports issued in 2013 had recommended classification changes, compared with 20% of reports issued in 2017 (Supplementary Data 3).
ACMG-AMP/ClinGen scheme comparison
ClinGen and original ACMG-AMP schemes were concordant in 35 proband reports (65%) and across 32 variants (74%). Of the 19 discrepant report classifications (representing 11 variants), a majority (79%) were clinically equivalent (between LP/P or between LB/B categories). The ClinGen scheme was more likely to achieve a “pathogenic” classification, upgrading six variants (14%) classified as LP by ACMG-AMP. In addition, four variants (9%) classified as a VUS per the original ACMG-AMP scheme were LP or P when classified by ClinGen criteria. Comparisons between ClinGen and ACMG-AMP schemes are summarized in Fig. 1c.
Differences between original ACMG-AMP and ClinGen classifications could be attributed to a small proportion of revised criteria (Supplementary Data 2). Use of gene-specific domain evidence (PM1) to assign an additional “moderate” criterion contributed to upgraded classifications in nine variants (82%). More precise and tiered definitions of variant prevalence in clinical cases (PS4) contributed to discordant classifications of eight variants (73%). The ClinGen scheme permitted downgrade of one variant due to a lower population allele frequency threshold for invoking the standalone benign criterion (BA1).
Impact of classification changes
Implications of proposed classification changes for cascade testing in family members were recorded by both number of genotyped individuals and number of individuals in whom testing might be of benefit on review of the family pedigree (where this was available; Table 4). Of the 19 proband reports with classification changes suggested by ClinGen criteria, 9 (47%) were issued in families where additional cascade or segregation testing had been undertaken. The total number of reports with suggested classification changes, including all probands and any cascade-tested family members, was 77 reports (41 positive for the reported variant, and 36 negative). The mean number of individuals genotyped per family, excluding probands, was 3.1 (range 0–40 individuals). In this group, six pedigrees were available, which identified an additional 38 first- and second-degree relatives potentially available for cascade testing (mean 6 individuals per family, range 0–12).
When stratified by classification changes that were clinically actionable (eight changes), four families had undergone cascade testing, representing 17 genotyped individuals (mean 2.1 individuals tested per family, range 1–4 individuals). Of these, ten were positive for the reported variant and seven were negative. Four of the variants with actionable changes were suggested downgrades (LP to VUS or LB; molecular diagnosis revoked), representing nine genotyped individuals (five testing positive and four negative for the reported variant). The remaining four variants in this group were upgrades from VUS to LP or P (molecular diagnosis confirmed), impacting four genotype-positive reports and an additional four genotype-negative reports. Only two pedigrees were available for review in this group, identifying 12 additional first- and second-degree relatives potentially available for cascade testing (mean 6 individuals per family, range 0–12).
DISCUSSION
Reclassification of previously reported MYH7 variants with the benefit of current information and best practice improves diagnostic confidence and simultaneously contributes to a dynamic evidence base that informs future classification. ClinGen’s modified MYH7 criteria provide a structured and gene-specific framework for interpretation of sequence variants that, in our cohort, produced a higher proportion of diagnostic reports and fewer VUS compared with standard ACMG-AMP guidelines. Comparison of evidence items selected across both schemes largely supports the rationale behind ClinGen’s revisions and provides insights into factors influencing classification differences.
Inherent uncertainty attached to “old reports” is anxiety-provoking for reporting scientists, clinicians, and their patients, especially when one considers the cascade clinical actions (and inactions) that result from declaration of a causative variant in a family. There is limited literature regarding the psychological impact of variant reclassification in inherited cardiomyopathy. Qualitative reports suggest that upgraded classifications are more likely to be met with relief (owing to higher confidence in the cause of the condition), while individuals with downgraded variants are more likely to experience worry, frustration, and disappointment at the loss of diagnostic security32. In addition, disclosure of a changed classification may impact decision-making in areas of reproductive planning, uptake of medical advice, lifestyle choices, and family communication32. Careful counseling, including discussion around uncertainty and the possibility of future reclassification, should occur in both the pre- and post-test settings.
In our cohort, the ClinGen MYH7-specific scheme resulted in a higher proportion of diagnostic (LP or P) classifications compared with original ACMG-AMP (65% vs. 54% of variants) and fewer VUS (30% vs. 42%). These findings are comparable with Mattivi et al., who demonstrated a nonsignificant reduction in VUS burden across two HCM cohorts23. The proportion of diagnostic classifications by variant did not differ considerably between original report (65%) and ClinGen schemes (65%), although notably these did not represent the same variants, as actionable changes were observed in 8 variants (18%) with a similar number of actionable upgrades (4) and downgrades (4). In this study, 39% of variants classified between 2013 and 2016 were reclassified compared with only 20% in 2017, suggesting that consideration be given to systematic review of MYH7 variants classified before 2017 with contemporary guidelines.
Scheme-specific differences contributed to classification changes in just over half of reports for both original ACMG-AMP and ClinGen schemes, suggesting that changes could not be accounted for by the emergence of new information alone. When new information contributed to reclassification by the ClinGen scheme, this was most frequently due to segregation data, clarification of functional/domain evidence, or expert group classification (each contributing to 30.8% of variant reclassifications). The latter two of these contributions flowed directly from the ClinGen initiative17, further highlighting the important role of expert group consensus in clarifying variant pathogenicity.
Comparisons of individual evidence items selected in both schemes provided further insights into gene-specific disease mechanisms and rationales behind ClinGen’s revisions (Table 2; Supplemental Data 2). In our cohort, PVS1 (null variant) was never selected per original ACMG-AMP criteria as all reported variants were missense or splice, consistent with the mutational spectrum of the condition and supporting removal of this criterion. The same can be said of removal of the PM3, BP1, and BP3 criteria (wrong mutational mechanism), and of PP4 (phenotype specificity) and BS2 (nonpenetrance), which do not apply in MYH7-associated cardiomyopathy. Conversely, ACMG-AMP permits selection of PP2 (missense variant in a gene with minimal benign missense variation) in every missense variant (93% of variants in our cohort), limiting its usefulness in discriminating between pathogenic and benign variation. Removal of this item did not have an impact on classification for any of the variants, further supporting its removal.
The revised item most commonly contributing to a classification difference was PM1 (functional/domain evidence). Clear definition of the requisite domain evidence in the ClinGen scheme allowed assignment of an additional “moderate” criterion in 28 variants (65%) and contributed to a classification change in 9 of these (21% of variants and 73% of those with classification changes). The unmodified ACMG-AMP PM1 criterion (“well-established functional domain”) might now take into account domain evidence asserted in the ClinGen paper; however, our laboratory did not apply PM1 prior to availability of the revised guidelines, and as our study aimed to evaluate the impact of ClinGen revisions this item was not selected in the ACMG-AMP scheme in order to facilitate meaningful assessment of the impact of ClinGen’s refined guidelines.
Several items in the revised ClinGen criteria have tiered weighting based on the volume of evidence available. The refined definition and tiered weighting of PS4 (prevalence in affected individuals) permitted additional assignment of pathogenicity criteria in 12 variants (28%) that would not otherwise have met the “strong” weighting by ACMG-AMP. This contributed to a classification change in eight variants (19%). Similarly, reweighted PP1 tiers (segregation evidence) allowed selection of an additional “strong” weighting in seven variants (16%) and “moderate” weighting in two variants (5%), but only contributed to an upgraded classification in two variants (5%). Our utilization of this criterion may have been limited by segregation testing being undertaken in only 26% of variants, thus the potential utility of the PP1 (and PS2; “de novo variant”) items may be underrepresented. Members of the curating multidisciplinary team also valued the clarity these tiered items provided over their unmodified ACMG-AMP equivalents, which were frequently prone to subjective interpretation.
The revised population evidence threshold (PM2) permitted an additional moderate criterion in two variants (5%) and was responsible for the upgrade of one variant (2%). The ClinGen working group has recently proposed a revision to reduce weighting of PM2 from moderate to supporting evidence31. While this recommendation was not available at the time of study curation, it is recognized that a reduced PM2 weighting would remove one moderate piece of evidence for multiple variants, and therefore potentially result in a downgraded classification. Further study could evaluate the impact of this new proposal. ClinGen’s revised benign criteria resulted in a classification downgrade in only one variant in our cohort (2%) compared with original ACMG-AMP criteria, owing to a lower permissible population allele frequency for selection of BA1.
We assessed the predicted clinical impact for the proband on the basis of whether a molecular diagnosis was confirmed where it was not previously, or was revoked. Classification by ClinGen resulted in an equal number of clinically actionable upgrades and downgrades, with actionable changes recommended in 18% of variants. The study cohort consisted of clinically affected individuals in whom diagnostic testing was undertaken. Seven individuals underwent testing due to sudden death, thus proband clinical utility was minimal in these cases. Report changes were recommended in three individuals with sudden death, and only one of these was an “actionable” change. In the surviving probands, molecular confirmation (or revocation) of the diagnosis was unlikely to modify medical management or surveillance, but might preclude (or enact) further investigation for a cause.
Projected cascade impacts for family members were more considerable, as predictive testing informs the need for echocardiographic surveillance. In our cohort, each classification change in a proband report impacted, on average, an additional 3.1 cascade reports for genotyped family members with a further 6.3 first- and second-degree relatives potentially available for cascade testing per family. The latter projection relies on a number of assumptions, including that “untested” relatives were still alive since documentation of the pedigree and that they had not undergone subsequent testing in another laboratory. Interpretation of clinical impact was also limited by the relatively small number of reports in which a clinical file and pedigree were available for review, especially when stratified for actionable changes. This study relied largely on information available at time of referral for testing, which in most cases was several years prior to study curation. Additional assessment of clinical impact might involve survey of referring clinicians and families with review of contemporary clinical history and pedigree information.
Few studies have examined the laboratory and clinical impact of variant reinterpretation in inherited cardiomyopathy. Laboratories typically reclassify variants in a reactive and ad hoc fashion, usually following a new request from a referring clinician or identification of a previously classified variant in a new individual. Systematic or periodic reinterpretation has been perceived as potentially unfeasible, costly, and logistically challenging for laboratory services, with the responsibility of initiating re-evaluation considered to lie with the patient and clinician27,33,34. Reclassification may also warrant report reissue, an undertaking that presents logistical challenges for the reporting laboratory, particularly if the original report was issued some time ago, and may not attract remuneration.
Periodic re-evaluation of variants, with the benefit of new information and improved understanding of the genetic architecture of HCM, poses logistical and economic challenges for laboratories but may have wider public health benefits. Further evaluation is needed to understand the potential social and health economic gains that may ensue from a refined molecular diagnosis within a family. These might include escape from surveillance of unaffected genotype-negative individuals or availability of cascade testing in families where an upgraded classification has occurred. ClinGen’s revised ACMG-AMP criteria provide clear expert-informed guidance for interpretation of sequence variants in MYH7 and a proof-of-concept that will undoubtedly presage future evidence-based revisions across a variety of genes and disease groups.
Data availability
Variant data and genomic coordinates are supplied in Supplementary Data 1 and are also available upon request to the corresponding authors.
References
Maron, B. J., Gardin, J. M., Flack, J. M., Gidding, S. S., Kurosaki, T. T. & Bild, D. E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 92, 785–789 (1995).
Maron, B. J., Maron, M. S. & Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J. Am. Coll. Cardiol. 60, 705–715 (2012).
Das, K. J., Ingles, J., Bagnall, R. D. & Semsarian, C. Determining pathogenicity of genetic variants in hypertrophic cardiomyopathy: importance of periodic reassessment. Genet. Med. 16, 286–293 (2014).
Cirino, A. L. & Ho, C. in GeneReviews (eds Adam, M. P. et al.). Hypertrophic cardiomyopathy overview. (University of Washington, Seattle, 1993).
Walsh, R. et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203 (2017).
Ho, C. Y. et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 138, 1387–1398 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Bean, L. J. H. & Hegde, M. R. Clinical implications and considerations for evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Med. 9, 111 (2017).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536, 285–291 (2016).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581, 434–443 (2020).
Karczewski, K. J. et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. Preprint at bioRxiv (2019) https://doi.org/10.1101/531210.
Ghosh, R., Harrison, S. M., Rehm, H. L., Plon, S. E. & Biesecker, L. G., ClinGen Sequence Variant Interpretation Working Group. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum. Mutat. 39, 1525–1530 (2018).
Abou Tayoun, A. N. et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524 (2018).
Amendola, L. M. et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am. J. Hum. Genet. 98, 1067–1076 (2016).
Nykamp, K. et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 19, 1105–1117 (2017).
Rivera-Munoz, E. A. et al. ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum. Mutat. 39, 1614–1622 (2018).
Kelly, M. A. et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 20, 351–359 (2018).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42(Database issue), D980–5 (2014).
Gelb, B. D. et al. ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet. Med. 20, 1334–1345 (2018).
Lee, K. et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum. Mutat. 39, 1553–1568 (2018).
Mester, J. L. et al. Gene-specific criteria for PTEN variant curation: recommendations from the ClinGen PTEN Expert Panel. Hum. Mutat. 39, 1581–1592 (2018).
Morales, A. et al. Variant interpretation for dilated cardiomyopathy: refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ. Genom. Precis. Med. 13, e002480 (2020).
Mattivi, C. L. et al. Clinical utility of a phenotype enhanced MYH7-specific variant classification framework in hypertrophic cardiomyopathy genetic testing. Circ. Genom. Precis. Med. 13, 453–459 (2020).
Lawal, T. A. et al. Disclosure of cardiac variants of uncertain significance results in an exome cohort. Clin. Genet. 93, 1022–1029 (2018).
Macklin, S. K., Jackson, J. L., Atwal, P. S. & Hines, S. L. Physician interpretation of variants of uncertain significance. Fam. Cancer. 18, 121–126 (2019).
Bennett, J. S. et al. Reclassification of variants of uncertain significance in children with inherited arrhythmia syndromes is predicted by clinical factors. Pediatr. Cardiol. 40, 1679–1687 (2019).
El Mecky, J. et al. Reinterpretation, reclassification, and its downstream effects: challenges for clinical laboratory geneticists. BMC Med. Genomics. 12, 170 (2019).
Richards, C. S. et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 10, 294–300 (2008).
Harris, P. A. et al. The REDCap consortium: building an international community of software platform partners. J. Biomed. Inform. 95, 103208 (2019).
Homburger, J. R. et al. Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc. Natl. Acad. Sci. U. S. A. 113, 6701–6706 (2016).
ClinGen. ClinGen sequence variant interpretation recommendation for PM2 version 1.0. https://clinicalgenome.org/working-groups/sequence-variant-interpretation/ (2020).
Wong, E. K. et al. Perceptions of genetic variant reclassification in patients with inherited cardiac disease. Eur. J. Hum. Genet. 27, 1134–1142 (2019).
Bombard, Y. et al. The responsibility to recontact research participants after reinterpretation of genetic and genomic research results. Am. J. Hum. Genet. 104, 578–595 (2019).
David, K. L. et al. Patient re-contact after revision of genomic test results: points to consider-a statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 21, 769–771 (2019).
Acknowledgements
The authors acknowledge and value the contributions of clinical and laboratory personnel involved in the care and diagnosis of these patients.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics Declaration
This project received Human Research Ethics Committee approval (HREC/52933/RCHM-2019) and site-specific approval through Murdoch Children’s Research Institute and Royal Children’s Hospital (Parkville, Victoria, Australia) in 2019.
Competing interests
C.M.R., T.Y.T., S.-J.P., B.C., S.L., and I.M. were salaried employees of the Victorian Clinical Genetics Services, part of the Murdoch Children’s Research Institute, Parkville, Victoria, Australia for the duration of this project. P.A.J. is a salaried employee of the Peter MacCallum Cancer Centre and Royal Melbourne Hospital, Parkville, Australia. There are no additional financial engagements or holdings to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Richmond, C.M., James, P.A., Pantaleo, SJ. et al. Clinical and laboratory reporting impact of ACMG-AMP and modified ClinGen variant classification frameworks in MYH7-related cardiomyopathy. Genet Med 23, 1108–1115 (2021). https://doi.org/10.1038/s41436-021-01107-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01107-y
This article is cited by
-
MYH7 in cardiomyopathy and skeletal muscle myopathy
Molecular and Cellular Biochemistry (2024)
-
Gendiagnostik bei kardiovaskulären Erkrankungen
Die Kardiologie (2023)
