
Abstract
Purpose
Neurofibromatosis type 1 (NF1) is associated with tumor predisposition and nonmalignant health conditions. Whether survivors of childhood cancer with NF1 are at increased risk for poor long-term health outcomes is unknown.
Methods
One hundred forty-seven 5+ year survivors of childhood glioma with NF1 from the Childhood Cancer Survivor Study were compared with 2629 non-NF1 glioma survivors and 5051 siblings for late mortality, chronic health conditions, and psychosocial, neurocognitive, and socioeconomic outcomes.
Results
Survivors with NF1 (age at diagnosis: 6.8 ± 4.8 years) had greater cumulative incidence of late mortality 30 years after diagnosis (46.3% [95% confidence interval: 23.9–62.2%]) compared with non-NF1 survivors (18.0% [16.1–20.0%]) and siblings (0.9% [0.6–1.2%]), largely due to subsequent neoplasms. Compared with survivors without NF1, those with NF1 had more severe/life-threatening chronic conditions at cohort entry (46.3% [38.1–54.4%] vs. 30.8% [29.1–32.6%]), but similar rates of new conditions during follow-up (rate ratio: 1.26 [0.90–1.77]). Survivors with NF1 were more likely to report psychosocial impairments, neurocognitive deficits, and socioeconomic difficulties compared with survivors without NF1.
Conclusions
Late mortality among glioma survivors with NF1 is twice that of other survivors, due largely to subsequent malignancies. Screening, prevention, and early intervention for chronic health conditions and psychosocial and neurocognitive deficits may reduce long-term morbidity in this vulnerable population.
Similar content being viewed by others

INTRODUCTION
Neurofibromatosis type 1 (NF1) is a common hereditary cancer predisposition syndrome affecting 1 in 2000 to 3000 children.1,2,3,4,5,6 Individuals with NF1 develop cancer at 2.7 times the rate found in the general population, often in childhood.7 Low grade gliomas are common childhood tumors in this population, and 15–20% of children with NF1 will develop an optic pathway glioma.8,9 In addition to elevated risk of benign and malignant tumors, children and adults with NF1 are predisposed to develop diverse chronic health conditions. NF1 frequently involves the skin and nervous system, resulting in cutaneous and plexiform neurofibroma.6 Neurocognitive deficits affecting attention, executive function and IQ are common, as well as behaviors similar to autistic spectrum disorder.10,11,12,13 Skeletal abnormalities, such as pseudoarthrosis, scoliosis, and increased fracture risk,14,15,16 as well as vascular complications including hypertension, arterial stenosis, and cerebrovascular abnormalities17,18 are also seen. Vision loss and pituitary dysfunction are also common complications of NF1 optic pathway gliomas.
Similarly, survivors of pediatric cancer are at increased risk for late mortality as well as adverse medical, mental health, and neurocognitive outcomes.19,20,21,22,23,24,25 Because children with NF1 are already at increased risk for difficulty in these domains, cancer therapies may further increase the risk of adverse late outcomes compared with survivors without NF1. The effects of NF1 on long-term outcomes in childhood cancer survivors is unknown. Measuring the risk of adverse late outcomes in survivors with NF1 may help guide early intervention efforts to reduce the impact of childhood cancer and its therapies in this population.
MATERIALS AND METHODS
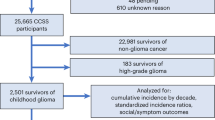
The Childhood Cancer Survivor Study (CCSS) is a multi-institutional, retrospective cohort with longitudinal follow-up of individuals who survived five years after diagnosis of cancer. Participants were less than 21 years old when diagnosed between 1 January 1970 and 31 December 1999 and responded to a CCSS questionnaire. Childhood cancer survivors with NF1 were either self-identified by reporting a personal history of NF1 or answering “yes” to the question “Have you ever been told by a doctor that you have neurofibromatosis (type 1)”. Survivors who responded “not sure” to this question but “yes” to the question “were you born with large or multiple birthmarks (any 1 larger than a quarter or 6 larger than a dime)” and had a primary cancer diagnosis associated with NF1 (including glioma, malignant nerve sheath tumor, rhabdomyosarcoma, or leukemia26,27) were also defined as NF1. Molecular diagnosis and family history were not considered in this analysis as only a limited number of survivors have had genetic testing and family history was incomplete in many survivors. Because glioma is the prevalent diagnosis among the NF1 survivors of childhood cancer, our primary analysis compared glioma survivors with and without NF1. A secondary analysis compared all survivors with NF1 to survivors without NF1 weighted to match primary cancer diagnosis. To determine whether survivors who were “not sure” of their NF1 status affected results, a sensitivity analysis examined late outcomes of NF1 glioma survivors who were self-identified.
The comparison of glioma survivors was performed in 2776 individuals (147 NF1 and 2629 non-NF1 survivors) and 5051 siblings of randomly sampled survivors. The comparison of all survivors included 22,492 survivors (176 NF1 and 22,316 non-NF1 survivors). Late mortality (death occurring >5 years from diagnosis) was ascertained from the National Death Index (NDI) through 2013, with causes of deaths predating the NDI (occurring 1975–1978) ascertained using state death certificates. Mortality was analyzed only for United States residents and only through 2013. Underlying causes of death for deceased participants according to the International Classification of Diseases, 9th and 10th Revisions (ICD-9 and ICD-10) were provided by the NDI. Causes of late mortality were grouped into categories of primary disease recurrence/progression, subsequent neoplasm, cardiopulmonary causes, external causes, and unknown/other causes.
For survivors diagnosed between 1970 and 1986, baseline questionnaires were completed between 1992 and 2004, and follow-up questionnaires during 2000–2003, 2002–2005, 2005–2006, 2007–2009, and 2014–2016. For survivors diagnosed between 1987 and 1999, questionnaires were administered between 2007 and 2014 (baseline) and 2014 and 2016 (follow-up). Details about specific questionnaires used in each outcome are summarized in Table S1 and available at (https://ccss.stjude.org/tools-and-documents/questionnaires.html).
Chronic health conditions were classified and severity-graded according to the Common Terminology Criteria for Adverse Events, version 4.0 (CTCAE v4.0). Late toxicities involving specific health conditions commonly involved in NF1 were also investigated (Table S2). Subsequent neoplasms occurring more than 5 years after initial cancer diagnosis were identified via self- or next-of-kin proxy report or death certificate and confirmed by pathology report or, when unavailable, death certificate, medical records, or both. Activities of daily living (ADL, including eating, bathing, dressing, or getting around the home) and instrumental activities of daily living (IADL, including household chores, necessary business, shopping, or getting around for other purposes) were self-reported. Self-reported history of any breast surgery (including biopsy, lumpectomy, or mastectomy), surgery for scoliosis, or placement of a ventriculoperitoneal shunt was also examined.
Emotional and behavioral problems were measured using the Brief Symptom Inventory 18 (BSI-18) for adult respondents and the Behavior Problem Index (BPI) for adolescents (<18 years of age). Neurocognitive outcomes were assessed for adult respondents with the CCSS Neurocognitive Questionnaire, which includes subscales measuring task efficiency, emotional regulation, organization, and memory. This scale was developed and validated in adult survivors of childhood cancer to assess neurocognitive impairment, including executive dysfunction.28 Self-reported learning and memory problems were also assessed using a single face-valid item. Emotional and neurocognitive impairment (excluding learning/memory) was defined as scores falling at or above the 90th percentile of sibling scores.
Socioeconomic outcomes included marital status, living status, employment, household income, and education. The first three outcomes were only measured in adults (≥18 years of age), while the others were evaluated in all available participants. Living dependently was defined as living with a parent, caregiver, or nondependent children and excluded living alone or with a spouse/partner. Unemployed participants were not working full- or part-time, and household income was dichotomized above or below $20,000 per year. Education was categorized as no college attendance or some college attendance with or without a college degree.
Demographic and treatment characteristics were summarized in survivors with and without NF1 and sibling groups. In analyses of all survivors not limited to glioma, survivors without NF1 were weighted to match the distribution of cancer diagnosis categories of survivors with NF1 to control for differences in diagnosis distribution. Late mortality was analyzed using Kaplan–Meier product-limit estimator for overall survival and cumulative incidence for cause-specific mortality, taking the other causes of death as competing risk events, comparing NF1, non-NF1 survivors, and siblings. Log-rank test of all-cause mortality curves and Gray’s test of cumulative incidence curves were applied to assess statistical significance of their differences. To adjust for the effects of differences in attained age, the mortality analysis comparing specific treatment subgroups was conducted using Cox proportional hazards models, adjusting for age at diagnosis as a covariate, in addition to using age as the time scale of the baseline hazard.
Multivariable regression approaches compared survivors with and without NF1 and siblings for nonmortality outcomes. Specifically, for emotional, neurocognitive, socioeconomic, service needs, and cancer-related pain outcomes, relative risk is reported using log-binomial regression. Piece-wise exponential regression models measured rate ratios for chronic health conditions, learning/memory problems, and medical procedures. Unanswered items were excluded from analysis. Since the NF1 group was relatively small, we applied the propensity score method for adjustments. Propensity scores were constructed by a multivariable logistic regression of the NF1 vs. non-NF1 outcome based on attained age, sex, race, diagnosis group, age at diagnosis, history of surgery, central nervous system radiation dose, and history of specific chemotherapy exposures (platinum, plant alkaloid, other chemo agent, intrathecal or intravenous methotrexate). Comparisons between NF1 survivors and siblings were adjusted for propensity scores that included attained age, sex, and race. Generalized estimating equations were used to account for potential within-family correlation between a survivor and his/her sibling of the same family. Statistical analyses were conducted using SAS version 9.4 (SAS Institute, Cary, NC) and R version 3.5.0. All statistical inferences were two-sided, and p values < 0.05 were considered statistically significant.
Ethics statement
The study was approved by the institutional review boards of the participating institutions, and informed consent was obtained from participants/guardians. A list of collaborating institutions can be found at https://ccss.stjude.org/learn-more/collaborating-institutions-and-principal-investigators.html.
RESULTS
Of 22,492 survivors in this study, 176 were determined to have NF1 and 147 (84%) of these had a primary cancer diagnosis of glioma. Among survivors with NF1, 153 (87%) were self-identified and 23 (13%) met the secondary definition of NF1. The mean (± standard deviation) of age at diagnosis of glioma survivors with and without NF1 was 6.8 (±4.8) years and 8.2 (±5.2) years, respectively. Mean follow-up was 15.0 (±4.9) years for glioma survivors with NF1 and 17.9 (±7.5) years for glioma survivors without NF1. Demographic and treatment characteristics are summarized by group in Table 1. Groups were similar in the proportion of baseline respondents that were able to be analyzed for each outcome (Table S3). Glioma survivors with NF1 were more commonly exposed to vincristine and platinum chemotherapy and less commonly exposed to radiation.
While all-cause late mortality among survivors remained similar up to 20 years from diagnosis (Fig. 1a, Table S4), mortality was subsequently increased among glioma survivors with NF1 (p = 0.01 for Kaplan–Meier comparison). For example, 30-year all-cause late mortality was 46.3% (95% confidence interval [CI]: 23.9–62.2%) in glioma survivors with NF1 vs. 18.0% (16.1–20.0%) in glioma survivors without NF1. While exposure to radiation or an alkylating agent led to increased mortality among glioma survivors without NF1 (p < 0.001), late mortality was similar between NF1 survivors with and without exposure to radiation and/or alkylating agents (p = 0.98, Fig. 1b). The most common causes of death among survivors with NF1 and glioma were subsequent neoplasms followed by unknown/other (Fig. 2). Among the ten deaths in glioma survivors with NF1 categorized as other, five were attributed to neurofibromatosis (ICD-10 code Q85.0), and three to neoplasm of uncertain behavior (D43.2).

(a) Late mortality in glioma survivors with neurofibromatosis type 1 (NF1), glioma survivors without NF1 and siblings. (b) Late mortality among glioma survivors by exposure to radiation or alkylator.

Cause-specific cumulative mortality starting five years from tumor diagnosis in glioma survivors with neurofibromatosis type 1 (NF1), glioma survivors without NF1, and siblings, for causes including subsequent neoplasm, recurrent/progressive disease, cardiopulmonary causes, external causes, and other/unknown.
Compared with survivors without NF1, CTCAE grade 3–4 chronic health conditions diagnosed before study entry (5 years after diagnosis) were more prevalent in glioma survivors with NF1 (46.3% [38.1–54.4] vs. 30.8% [29.1–32.6%], p < 0.001, Fig. S1). Glioma survivors with NF1 developed new grade 3–5 health conditions >5 years from diagnosis at a similar rate compared with glioma survivors without NF1 (rate ratio [95% CI]: 1.26 [0.90–1.77], Table 2, Table S5), although new grade 3–5 health conditions did arise at a greater rate among all survivors with NF1 compared with survivors without NF1 (1.53 [1.13–2.07]). Similarly, specific health conditions at study entry were more prevalent among survivors with NF1 than those without NF1 (Table S2). However, among glioma survivors, motor and sensory impairment, balance problems, and epilepsy were more common in survivors without NF1 while vision loss was more common among survivors of NF1-associated glioma. Compared with glioma survivors without NF1, glioma survivors with NF1 developed late-onset (>5 years from diagnosis) subsequent malignant neoplasm at four times the rate (4.02 [2.12–7.62]), but other NF1-specific health conditions were not significantly different between groups. The multivariable-adjusted rate of scoliosis repair surgery was over three times higher among glioma survivors with NF1 compared with glioma survivors without NF1 (3.81[1.81–8.03]), but there was no statistically significant difference in rates of procedures to place a ventriculoperitoneal shunt (0.66 [0.26–1.66]) or biopsy/resection of a breast lesion (0.83 [0.11–6.48]). A comparison of chronic health conditions in all survivors of childhood cancer also demonstrated increased risk of motor impairment (2.16 [1.33–3.52]) and epilepsy (2.05 [1.02–4.11]; Table 2, Table S5) associated with NF1.
Survivors of childhood glioma with NF1 who were adults at the time of evaluation had an increased risk of global distress (relative risk [95% CI]: 2.48 [1.25–4.91]) as well as depression (1.94 [1.01–3.76]), anxiety (3.22 [1.60–6.47]) and somatization (2.07 [1.05–4.09]) compared with glioma survivors without NF1. Compared with siblings, glioma survivors with NF1 showed an increased risk of emotional impairment on both the BSI-18 (all domains) and the BPI (peer conflict) (Table 3, Table S5). Compared with siblings, glioma survivors with NF1 had significantly increased risk of impairment in all neurocognitive domains, and increased risk of impaired task completion (1.50 [1.14–1.96]) compared with glioma survivors without NF1.
Glioma survivors with NF1 also showed increased risk of poor socioeconomic outcomes compared with both siblings and survivors without NF1 (Table 3, Table S5). Glioma survivors with NF1 were more likely to be unmarried (relative risk [95% CI]: 1.75 [1.05–2.94]) and not attend college (1.82 [1.18–2.78]) than survivors without NF1. Among all survivors, those with NF1 were also more likely to be unemployed (1.33 [1.05–1.69]), live dependently (1.41 [1.03–1.92]), have an annual household income ≤$20,000 (1.50 [1.01–2.23]), and require assistance with ADL or IADL compared with survivors without NF1. Restricting the definition of NF1 to those who self-identified yielded similar results (Table S6).
DISCUSSION
More than five years after diagnosis of a childhood glioma, survivors with NF1 experienced increased risk for late mortality, subsequent malignant neoplasm, neurocognitive deficits, and psychosocial impairment compared with survivors without NF1. Understanding these risks may help guide surveillance and early intervention efforts to improve outcomes in glioma survivors with NF1.
While all-cause late mortality among survivors remained similar up to 20 years from diagnosis, mortality rates beyond 20 years were subsequently increased among glioma survivors with NF1. By 30 years from diagnosis, late mortality for survivors with NF1 was more than twice that found in survivors without NF1 (up to 54% overall survival for glioma survivors with NF1). In comparison, population studies of individuals with NF1 have demonstrated increased mortality relative to the general population, but overall survival among individuals with NF1 in the general population remains >90% at 40 years of age.29
Subsequent neoplasms present one of the greatest risks of death to survivors with NF1. Due to their underlying pathologic variant, children with NF1 may be more likely to develop therapy-related second malignancies, and radiation therapy is avoided whenever possible.30,31,32 Studies of NF1 cohorts describe an increased risk of subsequent malignant neoplasms after radiotherapy but have not rigorously examined late mortality after a primary cancer diagnosis.31,32,33 As NF1 is a cancer predisposition syndrome, subsequent neoplasms may also develop independent of prior therapy. In our study, glioma survivors with NF1 developed a subsequent neoplasm at four times the rate of other glioma survivors, and subsequent neoplasm was the most common specific cause of death among survivors with NF1. Among survivors without NF1, exposure to radiation or alkylating agents was associated with increased late mortality, but a similar effect could not be shown among 54 NF1 subjects. Radiation therapy, although not alkylator exposure, has been associated with an increased risk of second neoplasm in NF1.34,35 However, despite changes in therapy to avoid DNA damaging therapies, survivors with NF1 may still be at increased risk for late mortality more than 20 years from their primary tumor diagnosis.
Because individuals with NF1 are at risk for nonmalignant health conditions,6,10,11,14,15,16,17,36 tumor therapies may increase the risk of chronic health conditions. In our study, survivors with NF1 start survivorship with a greater burden of medical conditions. Chronic health conditions are more prevalent among glioma survivors with NF1 than those without NF1 at cohort entry (5 years from diagnosis). However, new chronic health conditions (>5 years from diagnosis) develop at a similar rate between groups. Glioma survivors with NF1 therefore are at the same risk of developing new chronic health conditions as other survivors, and therapy was not associated with increased chronic health conditions in NF1. Among all cancer survivors (not limited to glioma), NF1 was associated with increased risk of late-onset (>5 years from diagnosis) severe/life-threatening chronic health conditions, as well as motor impairment and epilepsy among specific health conditions.
Findings of psychological distress and impaired task efficiency among survivors with NF1 are consistent with higher rates of attention and learning disorders in this population and support prior reports of social/emotional difficulties and executive dysfunction in young adults with NF1,10,37,38,39,40 though here in excess of what is typically associated with primary cancer therapy. Poorer socioeconomic outcomes suggest that survivors with NF1 may have fewer resources than non-NF1 survivors or siblings. Compared with survivors without NF1, survivors with NF1 were more likely not to have attended college, which is often associated with executive dysfunction, and were more likely to be unmarried. However, similar socioeconomic outcomes among NF1 individuals without a history of cancer are not available for comparison.
This study is subject to important limitations. Our study examines the effect of NF1 on childhood cancer survivors, but it is not able to measure the effect of childhood cancer on children with NF1. Late mortality in survivors with NF1 in our cohort appears significantly worse than published reports of mortality in the general NF1 population, but a direct comparison of late outcomes would require a comparison NF1 group without cancer is not available in the CCSS. Instead, this study examines the effect of NF1 in survivors of childhood cancer to guide surveillance and monitoring for late effects in clinics serving survivors with and without NF1.
The CCSS cohort provides a sample size large enough to make adjusted comparisons with non-NF1 survivors to isolate the independent effect of NF1 on late outcomes. However, there is a risk for misclassification as NF1 status was not confirmed by physical examination or genetic analysis. Inclusion criteria for NF1 may overlap with other syndromes, including constitutional mismatch repair deficiency (CMMRD), a rare childhood cancer susceptibility syndrome associated with neurocutaneous stigmata similar to NF1.41 The effect of this overlap is limited by the relative rarity of CMMRD (incidence of approximately 1:1,000,000 compared with 1:2000–1:3000 for NF1) and that CMMRD-associated brain tumors are frequently high-grade and are unlikely to survive five years from diagnosis to enter the cohort. We estimate that <0.5% of our NF1 cohort may have CMMRD, but a single survivor with CMMRD may affect rates of mortality due to subsequent malignancy.
In addition, all survivors with NF1 may not have been identified, so relative differences between survivors with and without NF1 may be underreported. Questions regarding NF1 status were posed differently in the baseline questionnaire than follow-up/expansion questionnaires (changing from “von Reckinghausen disease” to “neurofibromatosis type 1”), potentially contributing to differences in NF1 prevalence across decades. Improved awareness and ascertainment of NF1 status or decreased early mortality (occurring <5 years after diagnosis) over time may have also contributed to an increasing proportion of participants with NF1 over time. Similarly, racial and ethnic diversity of the NF1 cohort reflects the predominantly white, non-Hispanic background of CCSS survivors, but underrepresents NF1 survivors from other backgrounds. The effect of race and ethnicity, especially as it pertains to socioeconomic outcomes, is not able to be explored in this paper. The CCSS has attempted to maximize data quality and follow-up; however, questionnaire data rely on voluntary reporting. Self-report may overestimate or underestimate the severity of measured outcomes, and missing data due to unanswered questions may equally affect results in either direction. Within these limitations, however, we provide to our knowledge the largest study evaluating late outcomes in survivors of childhood cancer with NF1 with two important comparison populations to properly frame the rate of poor outcomes.
In conclusion, glioma survivors with NF1 have significantly higher rates of late mortality and subsequent neoplasm compared with glioma survivors without NF1. New chronic health conditions did not develop at a higher rate in glioma survivors with NF1, but they experienced poorer emotional, neurocognitive, and socioeconomic outcomes. Careful surveillance for common NF1-associated malignancies among survivors of childhood cancer with NF1 is warranted well into early adulthood. Differences in psychosocial and neurocognitive outcomes further highlight the need for additional surveillance and support for survivors of childhood cancer with NF1.
References
Li Y, Bollag G, Clark R, et al. Somatic mutations in the neurofibromatosis 1 gene in human tumors. Cell. 1992;69:275–281.
Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005;141:71–74.
Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–715.
Uusitalo E, Leppavirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015;135:904–906.
Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152A:327–332.
Huson SM, Compston DA, Clark P, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989;26:704–711.
Walker L, Thompson D, Easton D, et al. A prospective study of neurofibromatosis type 1 cancer incidence in the UK. Br J Cancer. 2006;95:233–238.
Prada CE, Hufnagel RB, Hummel TR, et al. The use of magnetic resonance imaging screening for optic pathway gliomas in children with neurofibromatosis type 1. J Pediatr. 2015;167:851–856. e851
Blanchard G, Lafforgue MP, Lion-Francois L, et al. Systematic MRI in NF1 children under six years of age for the diagnosis of optic pathway gliomas. Study and outcome of a French cohort. Eur J Paediatr Neurol. 2016;20:275–281.
Hyman SL, Shores A, North KN. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology. 2005;65:1037–1044.
Huijbregts SC, de Sonneville LM. Does cognitive impairment explain behavioral and social problems of children with neurofibromatosis type 1? Behav Genet. 2011;41:430–436.
Lehtonen A, Howie E, Trump D, Huson SM. Behaviour in children with neurofibromatosis type 1: cognition, executive function, attention, emotion, and social competence. Dev Med Child Neurol. 2013;55:111–125.
Chisholm AK, Anderson VA, Pride NA, Malarbi S, North KN, Payne JM. Social function and autism spectrum disorder in children and adults with neurofibromatosis type 1: a systematic review and meta-analysis. Neuropsychol Rev. 2018;28:317–340.
Gilbert A, Brockman R. Congenital pseudarthrosis of the tibia. Long-term followup of 29 cases treated by microvascular bone transfer. Clin Orthop Relat Res. 1995;314:37–44.
Crawford AH, Parikh S, Schorry EK, Von Stein D. The immature spine in type-1 neurofibromatosis. J Bone Joint Surg Am. 2007;89(Suppl 1):123–142.
Heerva E, Koffert A, Jokinen E, et al. A controlled register-based study of 460 neurofibromatosis 1 patients: increased fracture risk in children and adults over 41 years of age. J Bone Miner Res. 2012;27:2333–2337.
Rosser TL, Vezina G, Packer RJ. Cerebrovascular abnormalities in a population of children with neurofibromatosis type 1. Neurology. 2005;64:553–555.
Oderich GS, Sullivan TM, Bower TC, et al. Vascular abnormalities in patients with neurofibromatosis syndrome type I: clinical spectrum, management, and results. J Vasc Surg. 2007;46:475–484.
Gibson TM, Mostoufi-Moab S, Stratton KL, et al. Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970-99: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol. 2018;19:1590–1601.
Bhakta N, Liu Q, Ness KK, et al. The cumulative burden of surviving childhood cancer: an initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet. 2017;390:2569–2582.
Cheung YT, Brinkman TM, Li C, et al. Chronic health conditions and neurocognitive function in aging survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2018;110:411–419.
Zeltzer LK, Recklitis C, Buchbinder D, et al. Psychological status in childhood cancer survivors: a report from the Childhood Cancer Survivor Study. J Clin Oncol. 2009;27:2396–2404.
Gurney JG, Krull KR, Kadan-Lottick N, et al. Social outcomes in the Childhood Cancer Survivor Study cohort. J Clin Oncol. 2009;27:2390–2395.
Ellenberg L, Liu Q, Gioia G, et al. Neurocognitive status in long-term survivors of childhood CNS malignancies: a report from the Childhood Cancer Survivor Study. Neuropsychology. 2009;23:705–717.
Armstrong GT, Chen Y, Yasui Y, et al. Reduction in late mortality among 5-year survivors of childhood cancer. N Engl J Med. 2016;374:833–842.
Sung L, Anderson JR, Arndt C, Raney RB, Meyer WH, Pappo AS. Neurofibromatosis in children with rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma Study IV. J Pediatr. 2004;144:666–668.
Stiller CA, Chessells JM, Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer. 1994;70:969–972.
Krull KR, Gioia G, Ness KK, et al. Reliability and validity of the Childhood Cancer Survivor Study Neurocognitive Questionnaire. Cancer. 2008;113:2188–2197.
Evans DG, O’Hara C, Wilding A, et al. Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet. 2011;19:1187–1191.
Ater JL, Xia C, Mazewski CM, et al. Nonrandomized comparison of neurofibromatosis type 1 and non-neurofibromatosis type 1 children who received carboplatin and vincristine for progressive low-grade glioma: a report from the Children’s Oncology Group. Cancer. 2016;122:1928–1936.
Sharif S, Ferner R, Birch JM, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol. 2006;24:2570–2575.
Bhatia S, Chen Y, Wong FL. et al. Subsequent neoplasms after a primary tumor in individuals with neurofibromatosis type 1. J Clin Oncol. 2019;37:3050–3058.
Maris JM, Wiersma SR, Mahgoub N, et al. Monosomy 7 myelodysplastic syndrome and other second malignant neoplasms in children with neurofibromatosis type 1. Cancer. 1997;79:1438–1446.
Uusitalo E, Rantanen M, Kallionpaa RA, et al. Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol. 2016;34:1978–1986.
Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–314.
North KN, Riccardi V, Samango-Sprouse C, et al. Cognitive function and academic performance in neurofibromatosis. 1: consensus statement from the NF1 Cognitive Disorders Task Force. Neurology. 1997;48:1121–1127.
Hummelvoll G, Antonsen KM. Young adults’ experience of living with neurofibromatosis type 1. J Genet Couns. 2013;22:188–199.
Ejerskov C, Lasgaard M, Ostergaard JR. Teenagers and young adults with neurofibromatosis type 1 are more likely to experience loneliness than siblings without the illness. Acta Paediatr. 2015;104:604–609.
Graf A, Landolt MA, Mori AC, Boltshauser E. Quality of life and psychological adjustment in children and adolescents with neurofibromatosis type 1. J Pediatr. 2006;149:348–353.
Cohen JS, Levy HP, Sloan J, Dariotis J, Biesecker BB. Depression among adults with neurofibromatosis type 1: prevalence and impact on quality of life. Clin Genet. 2015;88:425–430.
Suerink M, Ripperger T, Messiaen L, et al. Constitutional mismatch repair deficiency as a differential diagnosis of neurofibromatosis type 1: consensus guidelines for testing a child without malignancy. J Med Genet. 2019;56:53–62.
Acknowledgements
This work was supported by grants from the National Institutes of Health (CA55727, and a Cancer Center Support (CORE) grant CA21765); the American Lebanese–Syrian Associated Charities (ALSAC); and the Neurofibromatosis Therapeutic Acceleration Program (Francis Collins Scholar to P.d.B.). This publication was supported by an agreement from the Johns Hopkins University School of Medicine and the Neurofibromatosis Therapeutic Acceleration Program. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of The Johns Hopkins School of Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
de Blank, P., Li, N., Fisher, M.J. et al. Late morbidity and mortality in adult survivors of childhood glioma with neurofibromatosis type 1: report from the Childhood Cancer Survivor Study. Genet Med 22, 1794–1802 (2020). https://doi.org/10.1038/s41436-020-0873-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0873-7
Keywords
This article is cited by
-
Temporal changes in treatment and late mortality and morbidity in adult survivors of childhood glioma: a report from the Childhood Cancer Survivor Study
Nature Cancer (2024)
-
Neurofibromatosis type1, type 2, tuberous sclerosis and Von Hippel-Lindau disease
Child's Nervous System (2023)
-
Comprehensively evaluating cancer survival in children with birth defects: a population-based assessment
Cancer Causes & Control (2022)
