
Abstract
Purpose
A primary barrier to improving exome sequencing diagnostic rates is the interpretation of variants of uncertain clinical significance. We aimed to determine the contribution of integrated untargeted metabolomics in the analysis of exome sequencing data by retrospective analysis of patients evaluated by both exome sequencing and untargeted metabolomics within the same clinical laboratory.
Methods
Exome sequencing and untargeted metabolomic data were collected and analyzed for 170 patients. Pathogenic variants, likely pathogenic variants, and variants of uncertain significance in genes associated with a biochemical phenotype were extracted. Metabolomic data were evaluated to determine if these variants resulted in biochemical abnormalities that could be used to support their interpretation using current American College of Genetics and Genomics (ACMG) guidelines.
Results
Metabolomic data contributed to the interpretation of variants in 74 individuals (43.5%) over 73 different genes. The data allowed for the reclassification of 9 variants as likely benign, 15 variants as likely pathogenic, and 3 variants as pathogenic. Metabolomic data confirmed a clinical diagnosis in 21 cases, for a diagnostic rate of 12.3% in this population.
Conclusion
Untargeted metabolomics can serve as a useful adjunct to exome sequencing by providing valuable functional data that may not otherwise be clinically available, resulting in improved variant classification.
Similar content being viewed by others


INTRODUCTION
The utilization of exome sequencing (ES) has substantially increased our capacity to identify disease-causing variants across a variety of clinical indications; however, ES has a variable diagnostic rate, and many patients remain without a clear diagnosis for a variety of reasons.1,2,3,4,5 Methods of supporting a definitive diagnosis remain a cardinal challenge for clinical ES, and a significant limitation is observed at the level of variant interpretation and classification. Though the American College of Medical Genetics and Genomics (ACMG), the Association for Molecular Pathology (AMP), and Clinical Genome Resource (ClinGen) have attempted to streamline and standardize this process through the publication and dissemination of official guidelines and various recommendations,6,7 there is ample evidence that interlaboratory variation continues to exist.8 Many of these discrepancies may be due to the lack of compelling and validated “functional” evidence as outlined in the ACMG framework. While in some cases such evidence can be easily obtained through complementary targeted biochemical testing or model organism studies, this is not always feasible or easily accomplished in a time-sensitive manner.
One approach to address this limitation would be to integrate untargeted metabolomic profiling with genomic analysis. Metabolomic profiling refers to the systematic identification and quantification of all substrates, intermediates, and metabolites in a given organism or biological sample at a point in time. In this way, a single test is able to assay multiple biochemical pathways at once and may lead to clear evidence of perturbations due solely to abnormalities in the variant in question. We have previously described the use of untargeted metabolomics in the screening and diagnosis of a variety of inborn errors of metabolism (IEMs)9 and the identification of novel disease biomarkers.10,11 We have also demonstrated that untargeted metabolomics may be useful in the diagnosis of individuals at either end of a phenotypic spectrum, as in the case of several individuals with adenylosuccinate lyase deficiency12 and in patients with GABA-transaminase deficiency.13 Here, metabolomic data were not only able to suggest a clinical diagnosis but also informed the choice of targeted molecular testing for additional confirmation. Untargeted metabolomics may, therefore, serve as a convenient and informative piece of strong functional data, and we have thus sought to use this combined approach in the analysis of a cohort of patient samples at our clinical laboratory.
MATERIALS AND METHODS
Sample and data acquisition
Clinical ES and global untargeted clinical metabolomic testing were ordered through a physician’s office or institution through standard procedures. Samples were sent to the Baylor Genetics Laboratory (Houston, TX; www.baylorgenetics.org) for processing and analysis. We surveyed our internal databases to identify cases where both metabolomic profiling and ES were requested for the same patient between May 2012 and August 2017. These studies were approved with a waiver of consent by the Baylor College of Medicine Institutional Review Board.
Untargeted metabolomic profiling
Metabolomic profiling (Global MAPS®) was performed by Baylor Genetics in collaboration with Metabolon, Inc. (Durham, NC) (www.metabolon.com), as described previously using two different platform configurations in plasma, urine, and/or cerebrospinal fluid (CSF).9,14 On the first platform configuration, samples were subjected to four chromatographic analyses: gas chromatography–mass spectrometry (GC-MS), liquid chromatography–tandem mass spectrometry (LC-MS/MS) in positive mode (LCMS Pos), LC-MS/MS in negative mode (LCMS Neg), and LC-MS/MS polar method (LCMS Pol). On the second platform, samples were subjected to the same mass spectrometry methods with the following chromatographic methods: LCMS Neg, LCMS Pol, and LCMS positive ion method focusing on lipophilic compounds (LCMS Pos Lipid) and LCMS positive ion method focusing on polar compounds (LCMS Pos Polar). The chemical structures of known metabolites were identified by matching the ions’ chromatographic retention index, nominal mass, and mass spectral fragmentation signatures with reference library entries created from authentic standard metabolites under the identical analytical procedure as the experimental samples. Raw spectral intensity values were normalized to the anchor samples, log transformed, and compared with a normal reference population to generate z-scores. Results were considered abnormally low if the z-score for a compound was equal or less than two standard deviations below the mean (i.e., ≤−2.00) or abnormally high if the z-score was equal or greater than two standard deviations above the mean (i.e., ≥2.00) of the control reference population.
Exome sequencing
ES data were acquired using previously described protocols1 developed by the Human Genome Sequencing Center at Baylor College of Medicine and adapted for clinical testing. Classification criteria for likely pathogenic (LP) and pathogenic (P) variants were based on the current ACMG/AMP guidelines, and a case was classified as molecularly diagnosed if pathogenic or likely pathogenic variants were detected in Mendelian disease genes that overlapped with described phenotypes of the patient in the appropriate inheritance pattern (i.e., biallelic for recessive disorders, etc.).3 Variant data were submitted to ClinVar, submission SUB7249674.
Integrated exome and metabolomics analyses
Integrated analysis of exome and metabolomic data was performed for each patient by extracting all pathogenic variants, likely pathogenic variants, and variants of uncertain significance (VUS) in genes associated with known inborn errors of metabolism (Supplementary Tables S1–S5).15 Untargeted metabolomic data were treated as functional evidence based on the ACMG and AMP guidelines for the interpretation of sequence variants (PS3/BS3). Metabolomic data were determined to confirm or upgrade a variant in the homozygous state or if the variant was in trans with another VUS, pathogenic, or likely pathogenic variant for an autosomal recessive disorder if there were characteristic analyte or metabolic pathway abnormalities. Metabolomic data supported the downgrade of a variant in the homozygous or hemizygous state if there were no abnormalities consistent with the IEM associated with that variant. The contribution of a variant was designated as ruled out in cases where there was no clear evidence of metabolic disturbances for a single VUS in the heterozygous state or in the case of compound heterozygous variants, and in these cases, no change in variant classification was made. Finally, metabolomic data were considered noncontributory or uninformative to variant interpretation if results were nonspecific, contradictory to a clear molecular or clinical diagnosis, if a subject had no variants in a known IEM, or if the assay was unable to detect the biochemical signature of the IEM in question.
RESULTS
Cohort characteristics and indications
Our cohort consisted of 170 patients referred to our diagnostic laboratory for ES and contemporaneous untargeted metabolomics between May 2012 and August 2017 (Supplementary Table S1). Patients were, on average, 7.61 years of age at the time of testing, with approximately equal sex distribution (56% male; 44% female). The majority of patients were referred for a primarily neurologic indication (91%). Of the 170 cases, 65 underwent trio ES (proband with both biological parents) while the remaining 105 underwent proband ES (with or without a parental control).
Integrated analyses
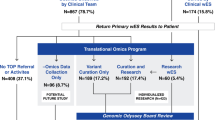
Of 170 patients, 74 (74/170 = 43.5%) were found to have at least one variant in an IEM gene amenable to assessment by our metabolomics platform (Fig. 1). These 74 individuals carried 145 variants in 73 different IEM genes, and metabolomic data contributed to the interpretation of 131 of these variants (131/145 = 90.3%) (Supplementary Table S2).

(a) Metabolomic data contributed to the interpretation of a variant in 44% of clinical cases. (b) In these cases, 74 individuals carried 145 variants amenable to interpretation via untargeted metabolomics. Of these variants, metabolomics helped to rule out the contribution of 60%, confirmed the pathogenicity of 11.7%, upgraded 12.4%, and downgraded 6.2%, according the American College of Medical Genetics and Genomics (ACMG) variant interpretation guidelines. LB likely benign, LP likely pathogenic, P pathogenic, VUS variant of uncertain significance.
For 87 (87/145 = 60.0%) single, heterozygous variants in autosomal recessive conditions, metabolomic data were able to rule out any contribution in terms of being a biochemically symptomatic carrier or possessing a second disease-causing allele not detected due to the constraints of ES.
Metabolomic data were able to contribute functional evidence to the interpretation of 42 (42/145 = 29%) identified variants, resulting in changes to their classification. Characteristic metabolomic profiles upgraded 18 variants (3 from LP to P and 15 from VUS to LP), while also downgrading 9 variants from VUS to likely benign (LB). In addition, profiles also revealed confirmatory perturbations for 17 (17/145 = 11.7%) pathogenic variants and therefore resulted in no classification changes. These assertions were based on identifying biochemical signatures in (1) autosomal recessive disorders with corresponding homozygous or compound heterozygous variants, (2) autosomal dominant disorders with corresponding heterozygous variants, and (3) X-linked disorders with corresponding hemizygous or heterozygous variants.
The variant reclassification due to metabolomic data resulted in confirmed diagnoses in 21 individuals based on clear biochemical evidence of an IEM and the identification of homozygous and/or compound heterozygous variants in autosomal recessive conditions, hemizygous variants in an X-linked condition, or single, heterozygous variants in autosomal dominant conditions. This approach resulted in an overall diagnostic rate of 12.3% (21/170 = 12.3% [Table 1]).10,13,16,17,18
DISCUSSION
With the increasing clinical use of ES and the implementation of clinical genome sequencing, there continues to be a need for tools capable of aiding in the interpretation of VUS and complex genetic backgrounds. While improvements in bioinformatics19 and functional assays20 have helped to alleviate some of this burden, there can still be significant limitations. Often these analyses result in imprecise and sometimes conflicting conclusions or require significant time to design and conduct an appropriate assay that may not have established clinical validity (i.e., research only testing). Of the 73 unique genes assayed by our metabolomic platform, for example, only 21 (21/73 = 28%) had readily available functional or confirmatory biochemical testing available in the United States (https://www.ncbi.nlm.nih.gov/gtr/).
Our results indicate that untargeted metabolomics can serve as a rapid and comprehensive way to screen VUS. The test has the advantage of being able to detect aberrations in hundreds of molecules indicative of many different metabolic pathways not easily assayed through standard biochemical assays. In this study, when assessed in conjunction with ES, untargeted metabolomic data were found to contribute to variant interpretation in 43.5% of cases (Fig. 1). For most of these variants, metabolomic profiling helped to rule out any likely clinical contribution, while in 22 cases, the data allowed for classification upgrade or confirmation of pathogenicity and diagnosis. Though our study population appears biased toward patients with neurologic disorders, this is consistent with previously reported pediatric referral indications to our clinical laboratory.1,3 Similarly, the majority of untargeted metabolomic samples were obtained from plasma, likely due to the ease with which this may be obtained, particularly in a pediatric population. Our results also highlight how useful metabolomic data can be in instances where an individual is found to carry only a single pathogenic or likely pathogenic variant in an IEM, either ruling out disease or supporting the need to look further for a second pathogenic variant. Caution should be taken, however, in the interpretation of variants associated with diseases in which patients may only intermittently manifest with metabolic abnormalities. It is well recognized, for example, that individuals with intermittent forms of maple syrup urine disease may exhibit normal biochemical markers outside of acute decompensations. For example, in the present study for patient 145 with biallelic variants in ALDOB consistent with hereditary fructose intolerance, metabolomic analysis showed only mildly elevated fructose, though it is possible that the patient had been treated at the time of sampling making it difficult to detect more characteristic abnormalities. This case highlights one important facet of metabolomic and other biochemical analyte analyses that should always be considered—the analysis reflects a patient’s biochemical features at a single point in time, reflecting diet, supplements, and medications at the time the sample is taken.
Our study, however, has some limitations. While our current metabolomic platform is able to detect the characteristic metabolic signatures of >100 inborn errors of metabolism, there are many disorders not currently reliably detectable by this method. These include mitochondrial respiratory complex deficiencies, glycogen storage disorders, most lysosomal storage disorders, and rare enzyme defects not previously assayed in our laboratory that require additional study and validation. Additionally, while untargeted metabolomic data in combination with exome/genome data may suggest or point to a specific diagnosis, in some cases, these findings may need to be confirmed via traditional assays, such as enzyme activity or assessment of additional body fluids, until associations are more clearly understood.
Our study also, however, highlights deficiencies in the way functional studies are treated in the current ACMG/AMP guidelines. At present, functional data may only be categorized as “PS3” or “BS3,” which is strongly supportive of either a variant’s pathogenicity or benign nature. Amended categorizations based on differing levels of support would be useful given the variable degrees of enzyme defects and the effects of treatment regimens or dietary interventions on biochemical phenotypes. Patient 1 (Table 1), for example, displayed minor abnormalities only modestly consistent with their molecular diagnoses, which may or may not be due to hypomorphic alleles or a specific treatment intervention. This stands in stark contrast to patient 92 where metabolomics identified the diagnostic signature10 of a peroxisomal disorder quite clearly. As the use of metabolomics continues to grow, there will be a need for additional ways of categorizing such data in a clear, standardized manner in the context of clinical outcomes.
Change history
01 August 2020
The original version of this Article contained an incomplete supplementary file in the Excel file format. The author has updated Table 2 in the file to include the ClinVar accession numbers. This has now been replaced in the HTML version of the Article.
References
Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–1511.
Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–1887.
Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879.
Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17:578–586.
Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18:696–704.
Rehm HL, Berg JS, Brooks LD, et al. ClinGen—the Clinical Genome Resource. N Engl J Med. 2015;372:2235–2242.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Amendola LM, Jarvik GP, Leo MC, et al. Performance of ACMG/AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98:1067–1076.
Miller MJ, Kennedy AD, Eckhart AD, et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J Inherit Metab Dis. 2015;38:1029–1039.
Wangler MF, Hubert L, Donti TR, et al. A metabolomic map of Zellweger spectrum disorders reveals novel disease biomarkers. Genet Med. 2018;20:1274–1283.
Burrage LC, Thistlethwaite L, Stroup BM, et al. Untargeted metabolomic profiling reveals multiple pathway perturbations and new clinical biomarkers in urea cycle disorders. Genet Med. 2019;21:1977–1986.
Donti TR, Cappuccio G, Hubert L, et al. Diagnosis of adenylosuccinate lyase deficiency by metabolomic profiling in plasma reveals a phenotypic spectrum. Mol Genet Metab Rep. 2016;8:61–66.
Kennedy AD, Pappan KL, Donti T, et al. 2-pyrrolidinone and succinimide as clinical screening biomarkers for GABA-transaminase deficiency: anti-seizure medications impact accurate diagnosis. Front Neurosci. 2019;13:394.
Ford LKA, Goodman KD, Pappan KL, Evans AM, Miller LAD, Wulff JE, et al. Precision of a clinical metabolomics profiling platform for use in the identification of inborn errors of metabolism. J Appl Lab Med. 2020;5:342–356.
Ferreira CR, van Karnebeek CDM, Vockley J, Blau N. A proposed nosology of inborn errors of metabolism. Genet Med. 2019;21:102–106.
Atwal PS, Donti TR, Cardon AL, et al. Aromatic L-amino acid decarboxylase deficiency diagnosed by clinical metabolomic profiling of plasma. Mol Genet Metab. 2015;115:91–94.
Bainbridge MN, Cooney E, Miller M, et al. Analyses of SLC13A5-epilepsy patients reveal perturbations of TCA cycle. Mol Genet Metab. 2017;121:314–319.
Soler-Alfonso C, Pillai N, Cooney E, Mysore KR, Boyer S, Scaglia F. L-Cysteine supplementation prevents liver transplantation in a patient with TRMU deficiency. Mol Genet Metab Rep. 2019;19:100453.
Lelieveld SH, Veltman JA, Gilissen C. Novel bioinformatic developments for exome sequencing. Hum Genet. 2016;135:603–614.
Starita LM, Ahituv N, Dunham MJ, et al. Variant interpretation: functional assays to the rescue. Am J Hum Genet. 2017;101:315–325.
Acknowledgements
The authors thank Leroy Hubert, Charul Gijavanekar, and Susan M. Toon for their assistance with data collection and helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
All authors were, at the time of this study, employees of Baylor College of Medicine, which has a partnership with Baylor Genetics that derives revenue from clinical testing. N.L. was supported by 5T32GM007526 Medical Genetics Research Program. K.E.G. was supported by the Takeda Pharmaceuticals/ACMG Foundation Next Generation Medical Biochemical Fellowship Award. V.R.S., S.H.E., and Y.Y. receive salary support from Baylor Genetics, a for-profit company that offers clinical testing referenced in this paper. V.R.S. is a paid advisory board member for PTC Therapeutics and CoA Therapeutics; he is also a noncompensated advisor for Mereo Biopharma. Y.Y. is the founder of AiLife Diagnostics, Inc., a for-profit company that provides interpretation and mining services. S.H.E.'s work is funded by the National Institutes of Health (NIH), Smith–Magenis Syndrome Research Foundation, PRISMS Inc., Shire Genetic Therapies, and Fondation Jerome Lejeune, and she is Chair of the Professional Advisory Board for PRISMS, and has received consulting fees from Enzyvant, Speragen, and Takeda. S.H.E. sits on the Board of the American Board of Medical Genetics and Genomics and the Board of the ACMG Foundation for Genetic and Genomic Medicine. V.R.S. sits on the Board of the Accreditation Council for Graduate Medical Education and the Board of the Society for Inherited Metabolic Disorders.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Alaimo, J.T., Glinton, K.E., Liu, N. et al. Integrated analysis of metabolomic profiling and exome data supplements sequence variant interpretation, classification, and diagnosis. Genet Med 22, 1560–1566 (2020). https://doi.org/10.1038/s41436-020-0827-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0827-0
Keywords
This article is cited by
-
The expanding diagnostic toolbox for rare genetic diseases
Nature Reviews Genetics (2024)
-
Clinical diagnosis of metabolic disorders using untargeted metabolomic profiling and disease-specific networks learned from profiling data
Scientific Reports (2022)
-
Diagnostic potential of the amniotic fluid cells transcriptome in deciphering mendelian disease: a proof-of-concept
npj Genomic Medicine (2022)
-
An integrated multiomic approach as an excellent tool for the diagnosis of metabolic diseases: our first 3720 patients
European Journal of Human Genetics (2022)
-
Assessment of the effects of repeated freeze thawing and extended bench top processing of plasma samples using untargeted metabolomics
Metabolomics (2021)
