
Abstract
Purpose
Enrichment of heterozygous missense and truncating SMAD6 variants was previously reported in nonsyndromic sagittal and metopic synostosis, and interaction of SMAD6 variants with a common polymorphism near BMP2 (rs1884302) was proposed to contribute to inconsistent penetrance. We determined the occurrence of SMAD6 variants in all types of craniosynostosis, evaluated the impact of different missense variants on SMAD6 function, and tested independently whether rs1884302 genotype significantly modifies the phenotype.
Methods
We performed resequencing of SMAD6 in 795 unsolved patients with any type of craniosynostosis and genotyped rs1884302 in SMAD6-positive individuals and relatives. We examined the inhibitory activity and stability of SMAD6 missense variants.
Results
We found 18 (2.3%) different rare damaging SMAD6 variants, with the highest prevalence in metopic synostosis (5.8%) and an 18.3-fold enrichment of loss-of-function variants comparedwith gnomAD data (P < 10−7). Combined with eight additional variants, ≥20/26 were transmitted from an unaffected parent but rs1884302 genotype did not predict phenotype.
Conclusion
Pathogenic SMAD6 variants substantially increase the risk of both nonsyndromic and syndromic presentations of craniosynostosis, especially metopic synostosis. Functional analysis is important to evaluate missense variants. Genotyping of rs1884302 is not clinically useful. Mechanisms to explain the remarkable diversity of phenotypes associated with SMAD6 variants remain obscure.
Similar content being viewed by others

INTRODUCTION
Craniosynostosis (CRS), the premature fusion of the cranial sutures, is a heterogeneous disorder with a prevalence of ∼1 in 2000. Environmental factors, polygenic inheritance and single-gene or chromosomal abnormalities all contribute to its complex manifestations. Variants in >60 genes have been identified as recurrently associated with CRS, with an underlying genetic cause being found in ∼24% of patients overall.1,2,3 The proportion in whom a cause can be determined varies widely depending on clinical diagnosis: from 88% for bicoronal synostosis down to 8% for sagittal synostosis (SS).2 Until recently, success in identifying a genetic diagnosis has been particularly low in nonsyndromic midline CRS, under 1% for both sagittal and metopic suture fusions.2
In 2016, Timberlake et al.4 performed exome sequencing of 132 parent–offspring trios and 59 additional probands presenting with clinically nonsyndromic SS, metopic (MS), or combined metopic/sagittal synostosis, seeking evidence for major monogenic contributions to these disorders. Based on enrichment of de novo variants and inherited damaging variants, this study identified a single significant gene, SMAD6, located at 15q22.3.4
SMAD6, originally identified in mammals by homology-based cloning,5,6 encodes one of two (with SMAD7) inhibitory members of the SMAD family required for regulated intracellular signal transduction by members of the transforming growth factor β/bone morphogenetic protein (TGFβ/BMP) superfamily.7,8,9 Intriguingly, enrichment of rare SMAD6 variants has also been reported in association with several other distinct phenotypes, namely congenital heart disease,10,11,12 bicuspid aortic valve (BAV) and ascending thoracic aortic aneurysm (TAA),13,14,15 intellectual disability,16 and radioulnar synostosis.17
In a follow-up study, Timberlake et al. increased the sample size of probands with midline CRS and no other genetic diagnosis to 379 (45 pedigrees included ≥1 additional affected family member).18 They found damaging SMAD6 variants in 4/234 (1.7%) SS, 11/135 (8.1%) MS, and 2/10 (20%) combined metopic/sagittal synostosis probands. Although de novo variants (DNMs) were identified in four families, in the remainder, the SMAD6 variant was transmitted by an apparently unaffected (i.e., nonpenetrant) parent. Similar observations of nonpenetrance of SMAD6 variants were made for several of the other described disease associations.13,14,17 To seek an explanation for the unpredictable penetrance, Timberlake et al.4,18 genotyped a single-nucleotide polymorphism (SNP), rs1884302, previously reported in a genome-wide association study (GWAS) of nonsyndromic SS to be the most significant associated SNP, which may differentially regulate the most proximal gene BMP2.19,20 The risk-conferring C allele (prevalence in non-Finnish Europeans of 32.7%, gnomAD),21 was found to be present in 15/21 individuals with CRS but only 1/20 unaffected relatives heterozygous for the SMAD6 variant but without CRS, suggesting a two-locus mechanism to account for variable manifestation of CRS.18
Although the studies described above4,18 represent an important advance in delineating the contribution of single-gene variants to nonsyndromic midline CRS, they raise several questions. First, what is the contribution of SMAD6 variants in all presentations of CRS (including syndromic diagnoses and fusion of coronal or lambdoid sutures)? Second, can it be assumed that all rare SMAD6 missense variants affect protein function? Third, can the two-locus (SMAD6/rs1884302) model be confirmed in an independent cohort? Here, we address these questions. We confirm the primary finding that SMAD6 variants are enriched in CRS, especially metopic synostosis, but find a more diverse pattern of clinical presentation; in addition, we illustrate the importance of combining functional studies with frequency-based evaluation of variants to refine likelihood of pathogenicity. Finally, we report that the two-locus model does not account for inconsistencies of penetrance of damaging SMAD6 variants in our data set.
MATERIALS AND METHODS
Patients
The clinical studies were approved by respective Institutional Review Boards (IRB): Oxfordshire Research Ethics Committee (REC) B (C02.143), London–Riverside REC (09/H0706/20), East of England–Cambridge South REC (14/EE/1112 for 100,000 Genomes Project [100kGP]), the Medical Ethical Committee of the Erasmus University Medical Center Rotterdam (MEC-2012–140), and the IRB of the University of California–Davis (International Craniosynostosis Consortium [ICC]; protocol 215635–23). Written informed consent to obtain samples for genetics research was given by each child’s parent or guardian. Written authorization for publication of clinical photographs was obtained from every individual and/or their parent/guardian. The clinical diagnosis of CRS was confirmed by three-dimensional computed tomography scanning of the skull; routine genetic investigations are described in Supplementary Information. Patients were considered to have a syndromic diagnosis if (1) additional dysmorphic features or congenital anomalies were present and/or (2) there was significant developmental delay or intellectual disability on neuropsychological assessment; in addition (3) families including affected first degree relatives were classified as syndromic, although affected individuals in those families may have presented with nonsyndromic clinical features.
Resequencing, bioinformatics, variant validation, and dosage analysis
Variant screening was performed by next-generation sequencing (NGS)-based resequencing of polymerase chain reaction (PCR) products encompassing the coding regions and intron/exon boundaries of the four exons of SMAD6 (NC_000015.10 [chr15:66702110–66782849, hg38]; NM_005585, ENST00000288840.9). Primer sequences are given in Table S1 and detailed methods are provided in Supplementary Information. Variant calls and coverage information were obtained using the bioinformatic tool amplimap.22
Validation and segregation analysis of variants was undertaken by dideoxy-sequencing of PCR products from genomic DNA. The rs1884302 BMP2 polymorphism was genotyped in all the SMAD6-positive individuals by PCR (Table S1) followed by EcoRI digest and/or dideoxy-sequencing.
Analysis of SMAD6 dosage using multiplex ligation-dependent probe amplification (MLPA) is described in the Supplementary Information.
Frequency- and deleteriousness-based variant stratification
Using the frequency-based filtering framework provided by Whiffin et al.,23 we estimated the maximum credible population allele frequency (AF) for any causative variant in CRS to be 0.000045, assuming a penetrance of 0.2 (Supplementary Information). For each identified variant we assigned AFmax, the greater of (1) the maximum observed AF in any population in gnomAD21 (except Other; gnomAD v2.1.1), where this was based on the presence of ≥2 mutant alleles in that population, or (2) the overall AF. We classified rare alleles as those for which AFmax was <0.000045.
To measure the deleteriousness of the identified variants, the deleterious score (DS, range 0–6) was calculated based on exceeding a defined threshold for six separate scores generated by Annovar24 (Supplementary Information).25 For comparison, the CADD score was additionally calculated for missense variants.26
For all SMAD6 variants listed in previous publications4,10,11,12,13,14,15,16,17,18 and implied to be pathogenic, nomenclature was verified using Mutalyzer27 and their presence and AF checked in gnomAD. We calculated the DS and CADD score for all missense variants.
Functional analysis
Source plasmids and methods used to evaluate the 5′ untranslated region (UTR), splice-site, and missense variants are provided in the Supplementary Information. Luciferase measurements and immunoblot quantifications were performed from at least three independent experiments. Statistical analysis is described in the Supplementary Information.
RESULTS
Resequencing of SMAD6 in 795 patients with CRS of unknown etiology
To investigate the contribution of SMAD6 variants in CRS, we performed NGS-based resequencing of SMAD6 in 795 unsolved patients with any type of CRS. After applying the joint criteria that variants should be both rare (AFmax < 0.000045) and damaging (predicted loss-of-function [LoF] and/or DS ≥ 4/6), we identified 18 probands (and 2 additional affected siblings) with heterozygous rare damaging SMAD6 variants (Table 1, Fig. 1, Fig. S1, Table S2). These SMAD6-positive individuals accounted for 2.3% of the cohort (3.4% and 2.0% of those classified as syndromic and nonsyndromic, respectively). Nine probands had LoF variants, representing a ~18.3-fold enrichment compared with gnomAD genome sequencing data (9 LoF in minimum 29,066 alleles; P < 10−7, Fisher’s exact test). The highest prevalence of novel/rare damaging SMAD6 variants occurred in MS (12/207; 5.8%). Such SMAD6 variants were much less frequent in isolated SS (3/316; 0.95%) or other types of suture fusion, but we noted three probands with coronal suture involvement (one each sagittal + bicoronal, sagittal + unicoronal, and unicoronal synostosis). The varied craniofacial presentation of individuals with CRS heterozygous for SMAD6 variants is illustrated in Fig. 2a–c.

Top, SMAD6 comprises four exons; positions of variants affecting translation initiation or splicing are indicated. Middle, cartoon of encoded protein showing conserved domains (MH1 and MH2, including highly conserved L3 region) and PY and PLDLS motifs.9 Colored shading indicates the position of the MH1 (transparent purple) and MH2 (transparent orange) domains according to Uniprot, Pfam, and CDD resources, with darker shading denoting overlapping domain assignments. Novel or rare (AFmax < 0.000045) variants identified in CRS patients that are also predicted damaging (loss-of-function [LoF], plus missense variants with DS25 ≥ 4) are indicated above the cartoon, whereas below in gray are additional missense variants predicted to have lower pathogenicity (DS ≤ 3 and/or AFmax > 0.000045); negative and positive controls10 used in the functional assays are colored green and blue, respectively. Frameshifts and stop-gain variants are shown with filled and empty arrowheads, respectively; § = de novo variants; * = novel/rare damaging variants found in addition to the CRS cohort screen; # = AFmax ≥ 0.000045 in gnomAD. Bottom, conservation profiles40 of inhibitory SMADs SMAD6 (black), SMAD7 (gray), and all SMAD members (SMAD1–8) combined (purple line). AF allele frequency, DS deleterious score.

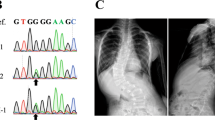
(a) Subject 8260 aged 4 months with metopic synostosis, the most frequently associated CRS phenotype, showing hypotelorism (front view) and trigonocephaly (top view). Newly described clinical presentations include sagittal and bicoronal synostosis (b, subject 3711 aged 11 months, note narrow, saddle-shaped skull with frontal bossing) and right unicoronal synostosis (c, subject 4370 aged 10 months, note facial asymmetry and recessed brow on right).
Together, these observations confirm the enrichment of rare, damaging SMAD6 variants in CRS, as reported in nonsyndromic midline synostosis.4,18 However, this work extends the previous findings to include syndromic as well as nonsyndromic patients, and synostosis of coronal sutures. Among the pure midline synostoses, MS was 6.1-fold more frequent than SS (P = 0.002, two-tailed Fisher’s exact test).
Phenotypic characterization of SMAD6-positive individuals
Combining these unbiased findings with eight additional independently identified (see Supplementary Information) SMAD6-positive CRS patients (three MS, three SS, one sagittal + bicoronal and one unicoronal synostosis; Fig. S2), in total we identified 25 different damaging variants in SMAD6 (Fig. 1, Table S2) in 28 affected individuals from 26 unrelated families with CRS.
The phenotypes identified in each of the 28 SMAD6-positive individuals are summarized in Table S3. Of 8 patients considered to have a syndromic clinical presentation, 1 (subject 5944) was excluded from further consideration owing to confounding by two additional DNMs likely to contribute to the phenotype, which had been separately identified by exome sequencing.28 Of the remaining seven syndromic subjects, five had congenital heart defects (comprising atrioventricular septal defect, atrial septal defect [ASD] with patent foramen ovale, two ASD with ventricular septal defect, and bicuspid aortic valve with right bundle branch block; only the first of these required corrective surgery), three had brain anomalies (comprising ventriculomegaly and absent corpus callosum, macrocephaly, and mild microcephaly), and one had duodenal atresia. Seven children had delayed developmental/intellectual or educational attainment, classified as mild–moderate in one case and mild in the remainder. Both congenital heart defects10,11,12 and neurodevelopmental disability16 were previously described as significantly associated with SMAD6 variants.
Although the craniofacial surgical course for most patients with CRS was good, four individuals (plus the child with additional confounding DNMs) were documented to have raised intracranial pressure (ICP). Of those four, one with syndromic SS developed additional bilateral coronal suture fusion and was found to have primary raised ICP and the other three (two SS, one MS) had raised ICP following reconstructive craniofacial surgery, necessitating a second major surgical procedure. Secondarily raised ICP represents an infrequent complication of simple synostosis of midline sutures, for example only 2 of 128 patients with SS,29 and 6 of 202 patients with MS30 who underwent major calvarial remodeling procedures developed secondarily raised ICP. The difference from background is significant (P = 0.042, Fisher’s exact test), suggesting that this is a complication that should be monitored for in SMAD6-positive CRS.
Mutational spectrum of SMAD6 and functional evaluation of the variants
The human SMAD6 protein comprises 496 amino acids and includes two highly conserved domains (MH1 and MH2), with MH2 being necessary and sufficient for its inhibitory activity on TGFβ/BMP signaling.31,32 The 25 different rare damaging variants in SMAD6 include 12 LoF (seven frameshift, three stop-gain, two splice-site), one within the 5′ UTR (creating an out-of-frame upstream ATG), an in-frame duplication (within the MH1 domain) and 11 missense variants (Fig. 1). Ten of the 11 identified missense variants are clustered in the MH1/MH2 domains and the remaining substitution is a de novo p.W14R located near the N-terminus and affecting a highly conserved region of the inhibitory SMADs (SMAD6/SMAD7; Fig. 1). We performed in vitro studies of the SMAD6 variants (excluding the frameshifts and stop-gains) to gather additional evidence for pathogenicity.
The two splice-site variants (c.817G>C and c.817+2T>A) are predicted to affect recognition of the intron 1 splice donor; in both cases, analysis of messenger RNA (mRNA) extracted from patient cells showed abnormal splice product(s), absent in the control (Fig. S3). Dideoxy-sequencing demonstrated activation of cryptic donor splice sites within intron 1, resulting in partial intron retention and generating a frameshift and premature stop codon (Fig. S3).
The 5′ UTR variant (c.-9G>T), present in both affected individuals of family M130 and assumed to have been inherited from their father (Fig. S1), creates an out-of-frame upstream ATG codon with potential initiation activity. Using a dual-luciferase reporter construct in C2C12 cells, the translation efficiency of the main open reading frame (ORF) was reduced, an effect not observed when the c.-9G>A SNP (rs559095945; AF[total]=0.0001706), affecting the same nucleotide but not generating an ATG codon, was tested (Fig. S4).
The inhibitory activity of the SMAD6 missense variants on BMP signaling was evaluated using a BRE-luc transcriptional reporter containing a BMP-responsive element.10,33,34 In addition to the 11 damaging and rare missense variants summarized in Table S2, we included 10 other missense variants that were either previously reported as positive (p.C484F) or negative (p.A325T) controls,10 or that we had encountered during resequencing (Table S2) but were classed as being of uncertain pathogenicity because they failed one or both criteria of being damaging and rare (Supplementary Information). SMAD6 missense variants located outside the MH2 domain maintained potent inhibitory activity, similar to the wild type (WT) and the p.A325T negative control (Fig. 3a). For SMAD6 variants located within the MH2 domain, the rare damaging variants associated with CRS (p.G365C, p.G390R, p.G473R, and p.E489K) showed reduced inhibitory capacity (statistically significant except for p.G365C), whereas more frequent and/or less damaging variants (p.S333T, p.K395R) did not. We found a stronger defect in variants within, or near to, the L3 loop motif, indispensable for the receptor association (Figs. 1 and 3a);9,35 the strongest defect was present for the p.G433fs construct, used as an additional control.

(a) Luciferase assay. The cartoon at top shows a simplified representation of the BMP signaling pathway, indicating in red the components transfected into C2C12 cells to perform the assay. Firefly luciferase activity of the BRE-luc transcriptional reporter induced by constitutively active BMPR1A (BMPR1A c.a.) was used to monitor the inhibitory effects of SMAD6 variants on BMP signaling, similar to previously described.10 Below, graphs represent means ± SEM from three independent experiments. Data were normalized (using Renilla levels), relativized to the wild type (WT) and analyzed by one-way analysis of variance (ANOVA) with Dunnett’s multiple-comparisons test; ∗P ≤ 0.05, ∗∗P ≤ 0.01, and ∗∗∗P ≤ 0.001. Color-coding of SMAD6 variants follows the same scheme as in Fig. 1. (b) Analysis of SMAD6 protein stability. Above are representative examples of western blots (using aliquots of protein extracts from the luciferase assays), showing SMAD6 protein levels (detected with anti-FLAG) compared with anti-GAPDH loading control. Control missense variants (negative, green bar, p.A325T; positive, blue bar, p.C484F) were selected as previously described.10 Data were normalized (using GAPDH) and relativized to the WT. The bars represent means ± SEM from three independent experiments, analyzed and visualized as in (a). (c) SMAD6 average protein levels of 21 different missense variants (from b) plotted against respective deleterious score (DS).
To test the stability of mutant SMAD6 proteins, we performed western blot analysis of the protein lysates generated for the BRE-luc reporter assay. Strikingly, all 11 CRS-associated rare damaging missense variants (and, in addition, a 5–amino acid in-frame duplication affecting the MH1 domain, and the C-terminal frameshift p.G433fs) exhibited significantly reduced SMAD6 protein levels, suggesting that the substitutions caused protein instability (Fig. 3b, red and pink bars). With the exception of p.E171K, average protein levels were <60% of WT. Conversely, no major instability was observed for eight other novel/rare variants predicted to have lower pathogenicity, or in more frequent (AFmax ≥ 0.000045) variants (Fig. 3b, gray bars). As an additional control we generated WT revertants of plasmids encoding three of the missense substitutions classified as pathogenic, and found that the revertant proteins were stable (Fig. S5).
Collectively, aside from the ten frameshifts/stop-gains (likely LoF alleles), we provide compelling functional evidence to support the pathogenicity of all 15 other CRS-associated rare and predicted damaging variants. The damaging missense variants mainly cluster in the MH1/MH2 domains of SMAD6 and significantly affect its activity and/or stability. We observed a strong negative correlation between the predicted deleteriousness of each missense variant and observed protein stability, with the DS (Spearman r = −0.67) (Fig. 3c) and CADD (Spearman r = −0.66) scores performing similarly (Fig. S6).
Re-evaluation of SMAD6 variants previously reported as pathogenic
An enrichment of SMAD6 variants considered to be pathogenic has been reported in several pathologies distinct from CRS.10,11,12,13,14,15,16,17 Using our variant categorization (based on AF and DS), we evaluated all SMAD6 variants that were previously reported as pathogenic. Systematic review identified 74 different SMAD6 variants, including 30 missense (Table S4, Fig. S7). Four of the 74 variants (including 3 missense) have an AFmax ≥ 0.000045; the in-frame deletion c.79_84del (p.S27_G28del, AFmax = 0.0007) is particularly frequent. This variant was identified in our initial resequencing (Table S2) but was excluded based on frequency and occurrence in a poorly conserved region outside the functional domains. Approximately one third of the previously reported SMAD6 missense variants are predicted to have a low (≤3) DS (Table S4, Fig. S8), affecting residues with low evolutionary conservation (Fig. S9).
Genotyping of BMP2 polymorphism (rs1884302) in SMAD6-positive individuals
Although 3 of the CRS-associated SMAD6 variants arose de novo, in 20 cases, the variant was transmitted from a clinically unaffected parent (11 mothers, 9 fathers); in 3 further cases, 1 or both parents were not available. The SMAD6-transmitting parents had a total of 23 children unaffected by CRS; taking account of the two affected sib pairs in the cohort, this indicates a sib recurrence risk of 2/25 = 8%, equivalent to an estimated penetrance for CRS of SMAD6 variants of ~16% assuming a 50% transmission rate of the parental SMAD6 variant (unaffected offspring were not genotyped).
To compare our results with the previous work of Timberlake et al.,4,18 which proposed that the rs1884302 SNP influenced the phenotype associated with SMAD6 variants, we genotyped rs1884302 in all SMAD6-positive individuals. However, we did not observe any association in our data between presence of the BMP2 risk allele (rs1884302C) and manifestation with CRS, whether the data were analyzed in a 2×2 association (Table 2) or by transmission disequilibrium test (in the 20 families with a SMAD6 carrier parent, parents heterozygous for the rs1884302 SNP transmitted 12 C and 7 T alleles to their affected offspring; P = 0.25, χ2 test). Merger of our 2×2 association data with the previous results of Timberlake et al.18 did however show a significant association (P = 0.002, Fisher’s exact test), although this was much weaker than observed in the original publication (Table 2).
DISCUSSION
This study builds upon the observations of Timberlake et al.4,18 to obtain a more comprehensive picture of the significance and impact of SMAD6 variants in CRS. We identified 28 affected individuals from 26 different families, adding to the 22 affected individuals from 18 families previously reported.18 By surveying all types of CRS without a genetic diagnosis, we gained a broader picture of the range of CRS phenotypes with which SMAD6 variants may be associated. We find that MS is most highly represented; 5.8% of patients with this diagnosis (Fig. 2a) had a rare, damaging SMAD6 variant, by far the largest monogenic contribution to MS yet identified.2 SMAD6 variants were less commonly associated with other types of suture fusion, in particular variants were significantly less frequent in SS (0.95% of cases overall) than in MS. We observed four SMAD6-positive patients in whom fusion of one or both coronal sutures accompanied the SS, as well as two patients with isolated unicoronal synostosis (Fig. 2b, c). Although based on small numbers, the observation that three subjects developed raised ICP following their primary surgical procedure suggests that long-term postoperative follow-up of these patients is important for optimal management. Given the inhibitory action of SMAD6 on TGFβ/BMP signaling, this work adds further evidence that overactivity of these pathway(s) predispose to craniosynostosis.1
Overall, we believe that the data presented here support adoption of SMAD6 genetic testing to inform genetic diagnosis of CRS: in the 13-year Oxford birth cohort study,2 10 of 677 (1.5%) individuals harbor rare, damaging SMAD6 variants, making SMAD6 the fifth most common gene for which variants are found in CRS within this cohort. The pattern of variants identified in patients with CRS (enrichment for heterozygous LoF, plus damaging missense), supports a haploinsufficiency mechanism of pathogenesis, predicting that partial or complete heterozygous deletions of SMAD6 would also be pathogenic. We screened for such lesions both experimentally (MLPA in 127 individuals with MS) and bioinformatically (113 individuals from 98 families with CRS), but did not detect any deletions in these samples or data sets (Supplementary Information).
The additional observation by Timberlake et al. that the genotype at the rs1884302 SNP appeared strongly related to manifestation of CRS in SMAD6-positive individuals4,18 has attracted attention as a potential example of digenic or two-locus inheritance.36 However, our own data do not support any major modifying role for this SNP (Table 2). Although upon merger with the previous data the overall effect remains significant (Table 2), we note that, given the frequency of the risk allele (C) of ~0.33 (gnomAD, European non-Finnish), the signal in these previous data4,18 was largely driven by strong underrepresentation of the C allele in nonpenetrant individuals, with only weak overrepresentation of C in penetrant individuals. This pattern runs counter to population genetic expectations, given the very low penetrance of CRS in individuals heterozygous for pathogenic SMAD6 variants (formally estimated as 0.16 from the number of additional affected and unaffected offspring born to carrier parents). Furthermore we note that the rs1884302 SNP was originally investigated because it showed the strongest relationship with nonsyndromic SS in a GWAS,19 but more recent data for MS reveal no equivalent association for this SNP.37 Given that the majority of individuals with deleterious SMAD6 variants have MS, it is perhaps unsurprising that we did not find an overall interaction between SMAD6 variants and rs1884302 genotype. Whether such a relationship might exist for the SMAD6-positive SS, and whether this interaction could be synergistic or simply represent an additive effect of the GWAS signal, will require a much larger sample size to answer. In conclusion, we caution that the modifying effect of rs1884302 on CRS phenotype in SMAD6 heterozygotes is unlikely to have useful predictive clinical application.
Despite the compelling statistical relationship between rare, damaging SMAD6 variation and CRS, many aspects of SMAD6 disease pathogenesis remain mysterious, a situation that poses substantial challenges for genetic counseling. Notably, although the spectrum of associated pathogenic variants shows the classical pattern of haploinsufficiency, the gnomAD pLI (probability of loss-of-function intolerance) value for SMAD6 is zero, as the number of LoF variants observed (23) exceeds expectation (11.7).21 Our own data support that many individuals carrying SMAD6 pathogenic variants remain asymptomatic, yet a minority are associated with significant morbidity. Besides the CRS by which these patients were ascertained, in eight cases syndromes were diagnosed based on concurrent congenital cardiac and/or significant neurodevelopmental disorders, features that have been shown to be associated with SMAD6 variants in independent studies.10,11,12,16
Substantial further work will be required to delineate the overall contribution of SMAD6 variants to multisystem pathogenesis, and to understand the causal mechanisms underlying the extreme variability in expressivity and the unpredictable penetrance. In this regard, for optimum practice it will be essential carefully to evaluate the pathogenic contribution of every SMAD6 variant encountered, because it is evident that there are many SMAD6 variants of intermediate rarity that may be found in mutational screens and assumed to be pathogenic, when this conclusion could be incorrect. Here, we show that in our CRS cohort, by applying joint thresholds for high predicted deleteriousness (DS ≥ 4) and low allele frequency (AFmax < 0.000045), we could successfully discriminate missense variants that demonstrated objective evidence of abnormal protein function in assays, based on overall stability (Fig. 3b, c) and (for those located in the MH2 domain), failure to suppress BMP-mediated signaling (Fig. 3a). In our analysis of previously reported missense variants implied to be pathogenic, 10/29 did not meet these joint criteria (Table S4). Such SMAD6 variants should be prioritized for further functional analysis to enable their clinical significance to be determined robustly, using logic similar to that recently described for CFTR variants.38
In determining the clinical utility of SMAD6 genetic testing, prior reports that subjects with BAV and ascending TAA are enriched for SMAD6 variation are particularly important, because these are occult cardiovascular disorders associated with severe age-related complications, potentially amenable to targeted echocardiographic screening and intervention. At present, however, the absolute and age-related risks of these disorders when association with SMAD6 variants are unknown. For families within the UK, we have started to offer echocardiographic screening to asymptomatic SMAD6-positive parents of children with CRS. Of the seven parents screened to date, none has shown evidence of either BAV or TAA, but unexpectedly, one had severe primary pulmonary hypertension (PPH). While this is not a described association of SMAD6 variants,39 two of the major known monogenic predispositions to PPH (variants in BMPR2 and SMAD9) also involve components of the BMP signaling pathway, so a pathogenic link to SMAD6 is plausible. These observations should motivate further efforts to disentangle the complex role of SMAD6 variation in multiple diseases.
Change history
07 July 2020
The HTML version of this Article previously published did not acknowledge Freya Boardman-Pretty and the Genomics England Research Consortium in the author list. This has now been corrected in both the PDF and HTML versions of the Article.
11 February 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41436-020-0886-2
References
Twigg SRF, Wilkie AOM. A genetic-pathophysiological framework for craniosynostosis. Am J Hum Genet. 2015;97:359–377.
Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr. 2017;29:622–628.
Goos JAC, Mathijssen IMJ. Genetic causes of craniosynostosis: an update. Mol Syndromol. 2019;10:6–23.
Timberlake AT, Choi J, Zaidi S, et al. Two locus inheritance of nonsyndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. eLife. 2016;5:e20125.
Riggins GJ, Thiagalingam S, Rozenblum E, et al. Mad-related genes in the human. Nat Genet. 1996;13:347–349.
Imamura T, Takase M, Nishihara A, et al. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389:622–626.
Hata A, Lagna G, Massague J, Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 1998;12:186–197.
Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol. 2016;12:203–221.
Miyazawa K, Miyazono K. Regulation of TGF-beta family signaling by inhibitory smads. Cold Spring Harb Perspect Biol. 2017;9:a022095.
Tan HL, Glen E, Topf A, et al. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum Mutat. 2012;33:720–727.
Jin SC, Homsy J, Zaidi S, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601.
Kloth K, Bierhals T, Johannsen J, et al. Biallelic variants in SMAD6 are associated with a complex cardiovascular phenotype. Hum Genet. 2019;138:625–634.
Gillis E, Kumar AA, Luyckx I, et al. Candidate gene resequencing in a large bicuspid aortic valve-associated thoracic aortic aneurysm cohort: SMAD6 as an important contributor. Front Physiol. 2017;8:400.
Luyckx I, MacCarrick G, Kempers M, et al. Confirmation of the role of pathogenic SMAD6 variants in bicuspid aortic valve-related aortopathy. Eur J Hum Genet. 2019;27:1044–1053.
Park JE, Park JS, Jang SY, et al. A novel SMAD6 variant in a patient with severely calcified bicuspid aortic valve and thoracic aortic aneurysm. Mol Genet Genomic Med. 2019;7:e620.
Lelieveld SH, Reijnders MR, Pfundt R, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–1196.
Yang Y, Zheng Y, Li W, et al. SMAD6 is frequently mutated in nonsyndromic radioulnar synostosis. Genet Med. 2019;21:2577–2585.
Timberlake AT, Furey CG, Choi J, et al. De novo mutations in inhibitors of Wnt, BMP, and Ras/ERK signaling pathways in nonsyndromic midline craniosynostosis. Proc Natl Acad Sci U S A. 2017;114:E7341–E7347.
Justice CM, Yagnik G, Kim Y, et al. A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal craniosynostosis near BMP2 and within BBS9. Nat Genet. 2012;44:1360–1364.
Justice CM, Kim J, Kim SD, et al. A variant associated with sagittal nonsyndromic craniosynostosis alters the regulatory function of a non-coding element. Am J Med Genet A. 2017;173:2893–2897.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Koelling N, Bernkopf M, Calpena E, et al. amplimap: a versatile tool to process and analyze targeted NGS data. Bioinformatics. 2019;35:5349–5350.
Whiffin N, Minikel E, Walsh R, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19:1151–1158.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Fu W, O’Connor TD, Jun G, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315.
Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13.
Burkardt DD, Zachariou A, Loveday C, et al. HIST1H1E heterozygous protein-truncating variants cause a recognizable syndrome with intellectual disability and distinctive facial gestalt: a study to clarify the HIST1H1E syndrome phenotype in 30 individuals. Am J Med Genet A. 2019;179:2049–2055.
Thomas GP, Johnson D, Byren JC, et al. The incidence of raised intracranial pressure in nonsyndromic sagittal craniosynostosis following primary surgery. J Neurosurg Pediatr. 2015;15:350–360.
Natghian H, Song M, Jayamohan J, et al. Long-term results in isolated metopic synostosis: the Oxford experience over 22 years. Plast Reconstr Surg. 2018;142:509e–515e.
Hanyu A, Ishidou Y, Ebisawa T, Shimanuki T, Imamura T, Miyazono K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J Cell Biol. 2001;155:1017–1027.
Nakayama T, Berg LK, Christian JL. Dissection of inhibitory Smad proteins: both N- and C-terminal domains are necessary for full activities of Xenopus Smad6 and Smad7. Mech Dev. 2001;100:251–262.
Goto K, Kamiya Y, Imamura T, Miyazono K, Miyazawa K. Selective inhibitory effects of Smad6 on bone morphogenetic protein type I receptors. J Biol Chem. 2007;282:20603–20611.
Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem. 2002;277:4883–4891.
Lo RS, Chen YG, Shi Y, Pavletich NP, Massague J. The L3 loop: a structural motif determining specific interactions between SMAD proteins and TGF-beta receptors. EMBO J. 1998;17:996–1005.
Komatsu Y, Mishina Y. An epistatic explanation. eLife. 2016;5:e21162.
Justice CM, Cuellar A, Bala K, et al. A genome-wide association study implicates the BMP7 locus as a risk factor for nonsyndromic metopic craniosynostosis. Hum Genet. 2020 Apr 7; https://doi.org/10.1007/s00439-020-02157-z [Epub ahead of print].
Raraigh KS, Han ST, Davis E, et al. Functional assays are essential for interpretation of missense variants associated with variable expressivity. Am J Hum Genet. 2018;102:1062–1077.
Zhu N, Pauciulo MW, Welch CL, et al. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 2019;11:69.
Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23:1875–1882.
Acknowledgements
We are very grateful to all the families for their participation in this study. We thank Deirdre Cilliers, Jacqueline Goos, Peter Noons, Rachel O’Keefe, Katie Rees, and Elizabeth Tidey for help with patient recruitment; Karen Crawford for curation of DNA samples; Hana Mlcochova and Sue Butler for cell culture; staff at the MRC Weatherall Institute of Molecular Medicine (MRC-WIMM) facilities for sequencing; Peter ten Dijke for plasmids; and Bart Loeys for discussions. This work was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme (A.O.M.W.), the National Institutes of Health–National Institute of Dental and Craniofacial Research (NIH-NIDCR) R01 DE-16886 (S.A.B.), Università Cattolica del Sacro Cuore linea-D1 2017–2018 intramural funds (W.L.), the MRC through the WIMM Strategic Alliance (G0902418 and MC UU 12025), the VTCT Foundation (S.R.F.T., A.O.M.W.) and a Wellcome Investigator Award 102731 (A.O.M.W.). We acknowledge support from the NIHR UK Rare Genetic Disease Research Consortium. This research was made possible, in part, through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the NIHR and National Health Service (NHS) England. Wellcome, Cancer Research UK, and the MRC have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the NHS as part of their care and support. The views expressed in this publication are those of the authors and not necessarily those of Wellcome, NIHR, NIDCR, or the Department of Health and Social Care.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
A.O.M.W. provides nonremunerated consultancy to Orion Corporation. The other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Calpena, E., Cuellar, A., Bala, K. et al. SMAD6 variants in craniosynostosis: genotype and phenotype evaluation. Genet Med 22, 1498–1506 (2020). https://doi.org/10.1038/s41436-020-0817-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0817-2
Keywords
This article is cited by
-
Whole genome sequencing identifies associations for nonsyndromic sagittal craniosynostosis with the intergenic region of BMP2 and noncoding RNA gene LINC01428
Scientific Reports (2024)
-
Clinical interest of molecular study in cases of isolated midline craniosynostosis
European Journal of Human Genetics (2023)
-
SMAD6 variants in nonsyndromic craniosynostosis
European Journal of Human Genetics (2023)
-
SMAD6-deficiency in human genetic disorders
npj Genomic Medicine (2022)
-
Benefits of clinical criteria and high-throughput sequencing for diagnosing children with syndromic craniosynostosis
European Journal of Human Genetics (2021)
