
Abstract
Purpose
Determine the variant detection rate for ENG, ACVRL1, and SMAD4 in individuals who meet consensus (Curaçao) criteria for the clinical diagnosis of hereditary hemorrhagic telangiectasia.
Methods
Review of HHT center database for individuals with three or more HHT diagnostic criteria, in whom molecular genetic analysis for ENG, ACVRL1, and SMAD4 had been performed.
Results
A variant known or suspected to be causal was detected in ENG in 67/152 (44.1%; 95% confidence interval [CI], 36.0–52.4%), ACVRL1 in 79/152 (52.0%; 95% CI, 43.7–60.1%), and SMAD4 in 2/152 (1.3%; 95% CI, 0.2–4.7%) family probands with definite HHT. Only 4/152 (2.6%; 95% CI, 0.7–6.6%) family probands did not have a variant in one of these genes.
Conclusion
Previous reports of the variant detection rate for ENG and ACVRL1 in HHT patients have come from laboratories, which receive samples from clinicians with a wide range of expertise in recognizing clinical manifestations of HHT. These studies suggest a significantly lower detection rate (~75–85%) than we have found in patients who meet strictly applied consensus criteria (96.1%). Analysis of SMAD4 adds an additional detection rate of 1.3%. HHT as defined by the Curaçao criteria is highly predictive of a causative variant in either ENG or ACVRL1.
Similar content being viewed by others

INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant vascular dysplasia that occurs in at least 1 in 10,000 individuals.1 It is characterized by small vascular lesions (telangiectases) of the oral cavity, lips, fingers, and mucosa of the nasal cavity and upper intestinal tract, as well as larger, fast flow vascular lesions (arteriovenous malformations [AVMs]) in the lungs, liver, and brain. Telangiectases and/or AVMs can occur elsewhere, but are not common or considered diagnostic of HHT.
HHT displays age-related penetrance and the average age for development and/or symptomatic presentation of telangiectases and AVMs is very organ specific. For example, recurrent nosebleeds (epistaxis) caused by bleeding telangiectases in the nasal mucosa is the most common symptom and eventually develops in more than 95%, but only 50% of diagnosed individuals report having nosebleeds by age 10.2 Oral/dermal telangiectases are typically not noted until the third decade of life.3,4 Thus, observable manifestations can be absent or subtle into adulthood. Yet, cerebral AVMs are usually congenital, with intracranial hemorrhage secondary to these lesions reported in infants and children with HHT.5 Since the diagnosis of HHT often cannot be made on clinical grounds in the first few decades of life, molecular diagnosis is key to implementation of medical management, which is recommended to begin in the first six months of life.6
HHT is a genetically heterogeneous disorder caused by pathogenic variants in multiple genes in the transforming growth factor beta (TGF-β) signaling pathway. Endoglin (ENG) and activin A receptor type II–like 1 (ACVRL1/ALK1) cause HHT1 (OMIM 187300), and HHT2 (OMIM 600376), respectively.7,8 Pathogenic variants in SMAD4 cause a combined juvenile polyposis/HHT (JP/HHT) syndrome (OMIM 175050).9 Pathogenic variants in these three genes lead to an underproduction of their respective proteins resulting in excessive, abnormal angiogenesis.10 HHT loci at chromosome 5q3111 and 7p14,12 identified by linkage analysis in one or two families respectively, have been designated as HHT3 and HHT4; however, over a decade later the genes remain unknown. Variants in GDF2 were identified in three individuals with clinical findings suspicious for HHT, but not meeting diagnostic criteria.13 In fact, more than two decades since the identification of ACVRL1 and ENG as genes associated with HHT, no new genes for HHT have been discovered.
Pathogenic variants in ENG and ACVRL1 account for roughly equal percentages of the disorder14 and are widely accepted, and often quoted, as causing a combined 75–85% of HHT based on multiple series reported in the mid-2000s.15,16,17,18,19 SMAD4 is reported to cause 1–2% of HHT.20 However, it is of note that these reports were based on series of cases submitted to diagnostic laboratories for clinical suspicion of HHT. Laboratories, including ours, receive samples from clinicians with a wide range of expertise in recognizing the clinical manifestations of HHT, as well as in understanding the consensus clinical diagnostic (Curaçao) criteria (Table 1).6,21
Methodology used in these mid-2000s series15,16,17,18,19 typically involved sequencing of exons and intron/exon borders and analysis for large deletions/duplications of ENG and ACVRL1. Our group has subsequently sequenced noncoding regions of these genes in cohorts selected from samples submitted to our laboratory for suspicion of HHT but with no pathogenic variant detected in the coding regions. Pathogenic variants outside the typically interrogated coding sequence were identified, particularly in the 5’UTR region of ENG and deep in intron 9 of ACVRL1.22,23 But these noncoding region variants explained a minority of all suspected HHT cases submitted to our clinical laboratory in which a pathogenic coding region variant of ENG or ACVRL1 had not been found.
Our group forms the core of the University of Utah HHT Center of Excellence, which has provided clinical diagnosis of HHT since 1995, and molecular genetic diagnosis in our Laboratory (ARUP Laboratories) since 2004. It has been our impression that the detection rate of causal variants in patients confirmed to have HHT based on family history, medical history, and physical examination at our specialty clinic is significantly higher than for all samples received into our laboratory from patients reported to have HHT. The purpose of this study was to assess the detection rate for a causal variant in ENG, ACVRL1, or SMAD4 for patients diagnosed with HHT based on a detailed, systematic clinical evaluation for manifestations of HHT and the strict application of the Curaçao diagnostic criteria.
MATERIALS AND METHODS
Methods consisted of review of the University of Utah HHT Center of Excellence patient database for individuals with three or more diagnostic criteria for HHT according to Curaçao criteria, and sequencing of exons and intron/exon borders for ENG, ACVRL1, deletion/duplication analysis of these genes if no suspicious variant was found by sequencing, then SMAD4 sequencing and deletion/duplication when no suspicious variant was detected in ENG or ACVRL1. Since 2011 sequencing of the 5’UTR region of ENG has also been included in our laboratory’s clinical testing protocol.22 One hundred fifty-two family probands (for genetic testing) were identified who met these criteria.
Clinical evaluation for all patients included history of nosebleed onset, as well as frequency, duration, and intensity; presentation of internal organ AVMs symptoms; careful examination for telangiectases at characteristic sites; and a targeted multigeneration pedigree. Screening for internal organ AVMs was routine for all those considered suspicious for, or confirmed with, HHT based on family history, medical history, and physical examination. The determination of affected status (three or more clinical criteria) for each individual was based on their complete evaluation, including internal organ screening. Molecular genetic testing of the ENG, ACVRL1, and SMAD4 genes has been recommended in all family probands with either suspected or definite HHT since clinical testing for these genes became available in the mid-2000s. In <20% of our clinic patients for whom genetic testing is recommended, it is not accomplished, most often due to lack of insurance coverage. This study was approved by the Utah Institutional Review Board (IRB 00039582).
RESULTS
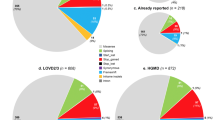
In patients who met Curaçao criteria, 96.1% (146/152; 95% confidence interval [CI], 91.6–98.5%) had a causal or likely causal variant in ENG or ACVRL1; an additional 1.3% (2/152; 95% CI, 0.2–4.7%) in SMAD4 (Fig. 1). Only 2.6% (4/152; 95% CI, 0.7–6.6%) did not have a variant in one of these genes.

Molecular genetic test results in 152 probands who met Curaçao criteria for hereditary hemorrhagic telangiectasia (HHT).
Of the 148 variants detected in these three genes, 141 individuals had a pathogenic or likely pathogenic variant. One hundred one of the pathogenic/likely pathogenic variants were found in a single proband and 14 were reoccurring (Table S1). The remaining seven variants were classified as variants of uncertain significance (VUS). A list of the VUS and evidence for pathogenicity is provided in Table 2. No other pathogenic variant or variant suspicious for being pathogenic was identified in any case.
DISCUSSION
Patients with a definite clinical diagnosis of HHT based on targeted physical examination, medical history, and family history had a causative variant in ENG, ACVRL1, or SMAD4 by sequencing of coding regions, intron/exon borders, and large deletion/duplication analysis approximately 97% of the time. This reaffirms the value of the Curaçao criteria in the clinical diagnosis of HHT, and also suggests that these known genes account for the vast majority of cases.
The 4 of 152 cases with no pathogenic or suspicious variant detected could represent patients with true HHT, with unidentified variants in a known gene or in an alternative unknown gene. It is of note that two of these four cases had additional molecular interrogation on a research basis; one had whole-genome sequencing and the other a custom genome sequencing test limited to the noncoding regions of known HHT/HHT overlapping genes. In neither case was a suspicious variant identified. For the other two cases, either the sample and/or consent was not available for additional testing on a research basis.
The alternative is that these 4/152 individuals were false positives for HHT by the application of Curaçao criteria. One had epistaxis, telangiectases, and a mother reportedly diagnosed with HHT but not examined by our team. Two had epistaxis, telangiectases, a single pulmonary AVM big enough to have been treated, but no family history except one daughter with epistaxis in one. The fourth has mild nosebleeds and relatively few telangiectases for age; but a sister who met diagnostic criteria (epistaxis, telangiectases, pulmonary AVM) and son with epistaxis and history of intracranial hemorrhage reportedly secondary to a cerebral AVM.
Overall, the high variant detection rate of ~97% for these three genes in this study suggests that many cases of presumed HHT found to be negative in previous laboratory based studies likely represent misapplication of the Curaçao criteria. In our laboratory, clinicians ordering genetic testing for HHT are contacted for various reasons to clarify clinical information provided on forms required with sample submission. Over time, the high frequency of misapplication of the Curaçao criteria, or lack of knowledge about the criteria, by physicians without familiarity with this rare vascular disorder has become apparent. For example, it is typical for red lesions on the trunk of the body most likely to be nonvascular pigmented lesions, to be noted as telangiectases. Or that epistaxis reported remotely in childhood, but not continuing into adulthood, is considered a diagnostic criteria. Or that an AVM of the extremity is considered suggestive of HHT. On one hand, a lower bar for clinical suspicion of HHT for purposes of ordering molecular genetic testing means that fewer cases of HHT will be missed. However, it has led to an underestimate of the variant detection rate in the known HHT genes in cases that meet clinical diagnostic criteria.
This underestimation of the clinical sensitivity of genetic testing for HHT has likely contributed to the underuse of molecular genetic testing in HHT families. This is concerning because the establishment of a causative variant in an HHT family allows for molecular diagnosis in at-risk family members. This is of particular importance in this disorder because it is not possible to rule out the diagnosis of HHT on clinical grounds in childhood; yet medical surveillance in the first six months of life is recommended according to consensus medical management guidelines. In particular, it is recommended that asymptomatic children of a parent with HHT be considered to have possible HHT, unless excluded by genetic testing. In addition, it is recommended that clinical screening for children with possible or definite HHT include magnetic resonance image (MRI) in the first six months of life to rule out a cerebral AVM.6 This examination is not trivial because it requires sedation at this age. It is thus medically indicated to rule out HHT by molecular genetic analysis in the first few months of life in those unaffected babies born to a patient with HHT, sparing them costly and unnecessary medical surveillance that should otherwise commence. Furthermore, a pathogenic variant in either ENG or ACVRL1 provides reassurance that the additional risks for gastrointestinal polyps and malignancy associated with a variant in SMAD4 is not a concern in a given family.
It should be noted that not all the sequence variants detected in the family probands in this study can be classified as pathogenic/likely pathogenic by current American College of Medical Genetics and Genomics (ACMG) guidelines.24 Missense variants are the most common variant type in both the ENG and ACVRL1 genes, and are difficult to classify as pathogenic by ACMG guidelines without functional studies. However, in our experience, most missense variants initially classified as VUS due to insufficient evidence of causation are actually pathogenic based on computational predictions and subsequent family cosegregation studies.25 Available evidence, provided in Table 2, suggests that these detected variants formally classified as VUS are more likely pathogenic than benign. Seven of seven of these variants were absent from gnomAD, indicating they are not common polymorphisms.
Despite the difficulty of interpreting noncoding region variants in general, we have recently shown the potential value of including a CT-rich hotspot region of ACVRL1 intron 9 and the 5’UTR of ENG in a molecular diagnostic testing algorithm for HHT.22,23 It is difficult to know how much this addition of noncoding region analysis will improve clinical sensitivity because the cohorts in which noncoding regions of HHT genes have been interrogated came from samples submitted to our laboratory with the previously mentioned limitation regarding inconsistency in applying HHT clinical diagnostic criteria. However, one case in this current series, with the pathogenic ENG c.−127C>T variant, would have been unexplained except for the addition of the ENG 5’UTR to our laboratory’s clinical testing algorithm a few years ago. On the other hand, more than two decades after the report of the ACVRL1 and ENG genes, no additional genes for HHT have been discovered.
In conclusion, for patients who have HHT according to the Curaçao diagnostic criteria, the detection rate is ~97% for a causative/likely causative variant in an exon or intron/exon border of ENG, ACVRL1, or SMAD4. This is much higher than previously reported in studies done using laboratory databases in which cases without definite HHT were included. This is encouraging because patients with this specific vascular malformation disorder are at risk for the serious consequences of AVMs in multiple internal organs, which can largely be prevented with early diagnosis and medical screening. Relatedly, as-yet undiscovered genes likely contribute little if any to the causation of HHT; and variants in noncoding regions of the known genes may explain the small percentage of HHT cases in which no pathogenic variant is currently identified.
References
Marchuk DA, Guttmacher AE, Penner JA, Ganguly P. Report on the workshop on hereditary hemorrhagic telangiectasia, July 10-11, 1997. Am J Med Genet. 1998;76:269–273.
Berg J, Porteous M, Reinhardt D, et al. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet. 2003;40:585–590.
Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. 1989;32:291–297.
Porteous ME, Burn J, Proctor SJ. Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet. 1992;29:527–530.
Morgan T, McDonald J, Anderson C, et al. Intracranial hemorrhage in infants and children with hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome). Pediatrics. 2002;109:E12.
Faughnan ME, Palda VA, Garcia-Tsao G, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48:73–87.
Shovlin CL, Hughes JM, Tuddenham EG, et al. A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nat Genet. 1994;6:205–209.
Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195.
Gallione CJ, Repetto GM, Legius E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363:852–859.
Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006;43:97–110.
Cole SG, Begbie ME, Wallace GM, Shovlin CL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42:577–582.
Bayrak-Toydemir P, McDonald J, Akarsu N, et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140:2155–2162.
Wooderchak-Donahue WL, McDonald J, O’Fallon B, et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet. 2013;93:530–537.
Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med. 2004;6:175–191.
Bossler AD, Richards J, George C, Godmilow L, Ganguly A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): correlation of genotype with phenotype. Hum Mutat. 2006;27:667–675.
Prigoda NL, Savas S, Abdalla SA, et al. Hereditary haemorrhagic telangiectasia: mutation detection, test sensitivity and novel mutations. J Med Genet. 2006;43:722–728.
Gedge F, McDonald J, Phansalkar A, et al. Clinical and analytical sensitivities in hereditary hemorrhagic telangiectasia testing and a report of de novo mutations. J Mol Diagn. 2007;9:258–265.
Richards-Yutz J, Grant K, Chao EC, Walther SE, Ganguly A. Update on molecular diagnosis of hereditary hemorrhagic telangiectasia. Hum Genet. 2010;128:61–77.
McDonald J, Damjanovich K, Millson A, et al. Molecular diagnosis in hereditary hemorrhagic telangiectasia: findings in a series tested simultaneously by sequencing and deletion/duplication analysis. Clin Genet. 2011;79:335–344.
Gallione CJ, Richards JA, Letteboer TG, et al. SMAD4 mutations found in unselected HHT patients. J Med Genet. 2006;43:793–797.
Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000;91:66–67.
Damjanovich K, Langa C, Blanco FJ, et al. 5’UTR mutations of ENG cause hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis. 2011;6:85.
Wooderchak-Donahue WL, McDonald J, Farrell A, et al. Genome sequencing reveals a deep intronic splicing ACVRL1 mutation hotspot in hereditary haemorrhagic telangiectasia. J Med Genet. 2018;55:824–830.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Bayrak-Toydemir P, McDonald J, Mao R, et al. Likelihood ratios to assess genetic evidence for clinical significance of uncertain variants: hereditary hemorrhagic telangiectasia as a model. Exp Mol Pathol. 2008;85:45–49.
Olivieri C, Pagella F, Semino L, et al. Analysis of ENG and ACVRL1 genes in 137 HHT Italian families identifies 76 different mutations (24 novel). Comparison with other European studies. J Hum Genet. 2007;52:820–829.
Finn RD, Attwood TK, Babbitt PC, et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017;45(D1):D190–D199.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121.
Acknowledgements
We thank the patients for participating in this research. We thank members of the ARUP Molecular Genetics and Genomics Clinical Laboratories for assisting in the sequence analysis of these patients. We thank the ARUP Institute for Clinical and Experimental Pathology for supporting this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
McDonald, J., Bayrak-Toydemir, P., DeMille, D. et al. Curaçao diagnostic criteria for hereditary hemorrhagic telangiectasia is highly predictive of a pathogenic variant in ENG or ACVRL1 (HHT1 and HHT2). Genet Med 22, 1201–1205 (2020). https://doi.org/10.1038/s41436-020-0775-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0775-8
Keywords
This article is cited by
-
Ruptured bilateral brain arteriovenous malformations in a young woman with early pregnancy: a case report
Journal of Medical Case Reports (2023)
-
ALK1 Deficiency Impairs the Wound-Healing Process and Increases Mortality in Murine Model of Myocardial Infarction
Journal of Cardiovascular Translational Research (2023)
-
Development and validation of a quality of life measurement scale specific to hereditary hemorrhagic telangiectasia: the QoL-HHT
Orphanet Journal of Rare Diseases (2022)
-
Imaging to intervention: a review of what the Interventionalist needs to Know about Hereditary Hemorrhagic Telangiectasia
CVIR Endovascular (2021)
