
Abstract
Purpose
Exome and genome sequencing (ES/GS) are performed frequently in patients with congenital anomalies, developmental delay, or intellectual disability (CA/DD/ID), but the impact of results from ES/GS on clinical management and patient outcomes is not well characterized. A systematic evidence review (SER) can support future evidence-based guideline development for use of ES/GS in this patient population.
Methods
We undertook an SER to identify primary literature from January 2007 to March 2019 describing health, clinical, reproductive, and psychosocial outcomes resulting from ES/GS in patients with CA/DD/ID. A narrative synthesis of results was performed.
Results
We retrieved 2654 publications for full-text review from 7178 articles. Only 167 articles met our inclusion criteria, and these were primarily case reports or small case series of fewer than 20 patients. The most frequently reported outcomes from ES/GS were changes to clinical management or reproductive decision-making. Two studies reported on the reduction of mortality or morbidity or impact on quality of life following ES/GS.
Conclusion
There is evidence that ES/GS for patients with CA/DD/ID informs clinical and reproductive decision-making, which could lead to improved outcomes for patients and their family members. Further research is needed to generate evidence regarding health outcomes to inform robust guidelines regarding ES/GS in the care of patients with CA/DD/ID.
Similar content being viewed by others


INTRODUCTION
Exome and genome sequencing (ES/GS) are relatively new clinical diagnostic genetic testing platforms for identifying a genetic etiology among individuals with congenital anomalies (CA), developmental delay (DD), or intellectual disability (ID). CAs are structural or functional abnormalities usually evident at birth, or shortly thereafter, and can be consequential to an individual’s life expectancy, health status, physical or social functioning, and typically require medical intervention. DD/ID are common features of a wide variety of genetic syndromes, or they could be isolated findings. Due to phenotypic and genetic heterogeneity associated with CA/DD/ID, establishing a syndromic diagnosis based on clinical signs and symptoms can be challenging, particularly in the newborn period. Furthermore, some CAs may not be easily diagnosed in the newborn period but could contribute to a lifelong burden to affected children and families. Clinical genetic testing can assist clinicians in confirming or establishing a clinical diagnosis that may lead to changes in clinical management, obviate the need for further testing, or end the diagnostic odyssey, which may improve outcomes for the patient and family.
Current standard practice, based on recommendations from the American College of Medical Genetics and Genomics (ACMG), is to perform chromosomal microarray (CMA) as the first-tier genetic test for individuals with CA/DD/ID.1 Diagnostic yield for CMA has typically focused on cohorts of mixed phenotypes of CA/DD/ID, and autism spectrum disorder (ASD). Yields averaged 12.2% in the early literature.2,3 More recent studies on cohorts of patients with CA/DD/ID have documented diagnostic yields ranging from 16% to 28%.4,5,6 As a result, CMA is a well-established tool in clinical practice, yet this testing will not capture single-nucleotide variations (SNVs) or small insertion/deletions (indels), smaller structural variants, and other pathogenic variant types contributing to CA/DD/ID.
The recommendations to perform CMA as a first-line test for CA/DD/ID occurred prior to the widespread availability of clinical ES/GS. Studies of ES/GS for these patients have shown there is an even larger diagnostic yield ranging from 28% to 68%.7,8,9,10,11,12 Notably, most studies included patients who had negative findings with CMA, demonstrating a higher yield for ES/GS compared with CMA.9 Higher yields have been observed when ES/GS is used as a first-line test.7,11
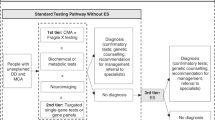
Establishing a diagnosis for patients with CA/DD/ID through genetic testing generally, and ES/GS specifically, can impact clinical management and patient-related outcomes (Figure 1). For example, early infantile epilepsy is a phenotypically and genetically heterogeneous disorder that may result from structural brain anomalies or functional abnormalities. Patients may be treated with diet, antiepileptics, or other systemic medications depending on their specific diagnosis; patients with West syndrome may be treated with adrenocorticotrophin, vigabatrin, or pyridoxine and biotin, while patients with Otahara syndrome who fail to respond adequately to antiepileptics may be treated with ketogenic diet or neurosurgery in the case of structural brain malformations.13

KQ key question.
The specific nature and frequency of outcomes describing clinical and personal utility resulting from ES/GS for patients with CA/DD/ID have not been well characterized; therefore, we initiated a systematic evidence review of the existing literature to document these. We focused our attention on the reported and demonstrated impact of ES/GS on clinical management, including anticipatory guidance, physical and social well-being, and the ability to influence reproductive decision-making for the patient or their family members. We summarize the extent and limitations of evidence for these outcomes in this patient population and include suggestions for prospective evidence generation to inform both clinical and personal utility resulting from ES/GS.
MATERIALS AND METHODS
In 2017, this ACMG working group was established to assess patient and clinical outcomes of ES/GS in patients with CA/DD/ID. Workgroup participants were members of ACMG and included board-certified medical geneticists specializing in adult and pediatric clinical genetics (L.D., J.G., S.E.H., D.T.M., E.M.P., A.C.-H.T., M.T.S.) and laboratory genetics (J.S., M.C.S.), and a methodologist (J.M.). None of the working group members had any conflicts of interest, according to ACMG policy. To address the overarching research question, “What is the utility of exome/genome sequencing of patients with CA/DD/ID?” the authors developed a description of the targeted population, intervention, comparator, outcomes, timing, and setting (PICOTS) (Table 1). In response to the PICOTS, key questions (KQs) were developed to structure and inform the overall goals of the project (Table 1). The KQs corresponded to the health outcomes, clinical management, reproductive planning/issues, and health-care utilization from the patient, family, and provider perspectives.
Search strategy
The following search strategy was used to query the PubMed database from 1 January 2007 to 26 April 2018 and was updated on 1 March 2019, with English and Human filters:
(((“whole exome” OR “whole genome”) AND sequencing) OR (WES) OR (WGS)) AND (clinical OR utility OR outcome OR treatment OR care OR medication OR intervention OR counseling OR stress OR relief OR depression OR anxiety OR psychological OR communication OR “quality of life” OR quality OR pregnancy OR prenatal OR carrier OR testing OR test OR reproductive OR anxiety OR decision OR referral OR surgery OR procedure OR management OR outcome)) NOT (bacterial OR microbial OR isolate OR isolates OR virulence OR infection)
Each publication identified by the search strategy underwent dual review for eligibility by a team of nine reviewers in a two-step process: title/abstract screening followed by full-text review using the inclusion and exclusion criteria listed in Table 1. Although CA may manifest postinfancy, we limited inclusion of studies to those where CA had onset prior to age 1 year and DD/ID had onset prior to age 18 years. Studies that failed to meet inclusion criteria were excluded in title/abstract review or in full-text review. Discordance between reviewers at each step was resolved through discussion, with adjudication by the methodologist or other group members, if needed. We identified additional publications through review of the references cited by included studies. We specifically sought to identify gray literature; that is, relevant data captured in laboratory and/or hospital registries of patients with CA/DD/ID, presented at conferences, and/or published as conference abstracts. Studies presenting only hypothetical impacts to patient or family management stemming from ES/GS, or studies that presented only the diagnostic yield of ES/GS were excluded.
Systematic Review Data Repository
The Systematic Review Data Repository (SRDR) (https://srdr.ahrq.gov/), a freely available web-based platform for data extraction and management of systematic evidence reviews was used to facilitate the evidence review. Individual SRDR projects were created for the title and abstract screening, full-text review, and data extraction of studies. Studies were uploaded in duplicate to SRDR and randomly assigned to reviewers for each stage of the evidence review. Extraction forms were developed separately. Specifically, the extracted data included study design details, sequencing method, patient population information (i.e., CA/DD/ID, age), and outcomes related to ES/GS, including harms associated with their use. Diagnostic yield was not extracted nor evaluated for this study.
Patient and clinical outcomes
We assessed the extent to which studies reported a measurable impact on health, reproductive, psychological, and behavioral outcomes for the patient or the patient’s family, including:
-
1.
Morbidity, mortality, change in health status or function, quality of life, or duration of hospitalization
-
2.
Reproductive decision-making for the patient or their immediate family
-
3.
Change in clinical management such as recommending medication/nutritional supplementation, ordering diagnostic tests or procedures, and referring to specialists
-
4.
Psychosocial and behavioral outcomes such as distress, anxiety or depression, or change in communication patterns among the patient and their family/caregivers
Data analysis
Extracted data were summarized in several ways. Counts were calculated for study characteristics (e.g., country, year of publication) for publications with 20 or more patients. We extracted evidence of outcomes of interest from each study and calculated the proportion of unique patients (or the family of a patient) with a reported change in management or reproductive planning. A narrative synthesis was performed due to substantial heterogeneity between studies and because available data precluded quantitative analysis.
RESULTS
The database searches identified 7178 publications. During title and abstract screening, 5405 studies were excluded based on the inclusion/exclusion criteria (Figure 2, Table 2). Additional studies, mostly case reports, were identified through exhaustive review of citations in other articles and conference abstract searches (n = 881); a total of 2654 publications were retrieved for full-text review. Following review of eligibility, 177 studies were selected for data extraction. During the data extraction process, 10 additional studies were removed. Outreach to hospital and clinical laboratories, and authors of relevant meeting presentations and abstracts, resulted in the inclusion of data from one additional source.14 In sum, 167 relevant studies, including case reports, were identified.

CA congenital anomalies, DD/ID developmental delay/intellectual disability.
Study characteristics
The majority of included studies were case reports or case series with small populations (n < 20 patients). Table 2 summarizes the study characteristics of the 36 included publications with a patient population ≥20. Sample size reported corresponds only to the number of patients in each study reported to have received ES/GS. In one study,15 the patient population was reported as the number of families that participated. For the 36 studies characterized in Table 3, the sample size ranged from 22 to 278 patients. Most studies reported ES results (n = 27), while 7 studies reported health or clinical management outcomes following GS; two studies used both methods. Studies were set predominantly in the United States (n = 15), but overall, reflected the growing international use of sequencing technology in patients with CA/DD/ID.
Outcomes of ES/GS for CA or DD/ID
We assessed changes in clinical management and patient outcomes resulting from ES or GS in recognition of the potential impact(s) a genetic diagnosis may have on the patient and their family. Of the 167 included studies, 95% reported a change to patient or family clinical management and most (79%) were case reports or case series with a small number of patients. Table 3 presents an overview of the 36 studies with N ≥ 20 patients and the reported health and clinical outcomes. Several studies provided only representative case examples documenting outcomes of interest.16,17,18,19 We summarize the number of studies with N ≥ 20 patients documenting the outcomes of interest and representative examples of reported health outcomes and clinical impacts for each subcategory.
Health outcomes
Mortality and morbidity were reported in three studies. In a case series of patients with CA, a patient in the pediatric intensive care unit (PICU) with suspected biliary atresia was scheduled for interoperative cholangiogram with reflex to Kasai hepatoportoenterostomy.20 A provisional diagnosis of Alagille syndrome obtained with GS (later confirmed by CMA) was conveyed to doctors urgently, as the patient was in the operating room for induction of general anesthesia for the planned procedure. In patients with Alagille syndrome, the Kasai procedure is associated with increased risk for liver transplant and higher mortality.21 The consensus of an international expert panel was that cancellation of the surgery led to the reduction in mortality risk by 83–94%, compared with the clinical course of a (nonstudy) patient who presented similarly and underwent the surgery the following month. In the same case series, morbidity was avoided in 61% (11/18) of patients who received a diagnosis with rapid GS (results returned in less than 2 weeks), compared with none of the patients who received standard of care,20 suggesting that obtaining a timely diagnosis may result in appropriate management and improved clinical outcomes. In a series of acutely ill infants in the neonatal and pediatric intensive care units (NICU and PICU), Willig et al. reported a higher 120-day mortality rate in 57% (12/21) of patients who received a genetic diagnosis with rapid GS compared with 14% (2/14) of patients who did not, suggesting this population is more medically fragile and may benefit from obtaining results quickly to guide care. However, this difference was not statistically significant (p = 0.46).22 A case series of patients undergoing rapid ES in Australia reported a mortality rate of 23% (9/40).23 Generally, however, reported health outcomes were often subjective in nature.
Clinical management outcomes
Baldridge et al. determined that 5% (8/155) of patients had their clinical management directly influenced as a result of ES/GS, including a change in medication, surgical procedures, or referral to specialists.24 Additionally, 54% (84/155) of these patients received additional diagnostic (i.e., molecular, biochemical, imaging) studies following ES, including 12 that were performed after the discovery of a secondary finding (i.e., ACMG59).24 Secondary findings were also reported in a case series of patients with CA/DD/ID.25 Tammimies et al. reported 6% (6/95) of patients had ES findings considered “medically actionable,” including identification of pathogenic variants in FBN1 (Marfan syndrome, familial thoracic aortic aneurysms and dissections), CACNA1S (malignant hyperthermia), and SDHB (hereditary paraganglioma–pheochromocytoma syndrome).25
Medication and dietary management
A change of patient medication, either initiation of a new treatment or halting of an existing one, was specifically reported in 22 studies.8,15,16,17,18,20,22,23,26,27,28,29,30,31,32,33,34,35,36,37,38,39 For example, Anazi et al. reported a patient with ID who was homozygous for a truncating pathogenic variant in SLC39A14 that causes a potentially treatable form of hypermanganesemia, who was started on chelation therapy, resulting in improved manganese levels.17 In a case series of patients with presumed neurogenetic disorders and DD/ID from Argentina, 10% (4/40) underwent a trial of new medication following ES, including a trial of L-Dopa in a patient with paraplegia and ID who had pathogenic variants in SPG11 and a trial of acetazolamide and fampridine in a patient with a pathogenic variant in KCNA2.39 In the same study, physicians made recommendations to avoid statins in a patient with a pathogenic variant in DMD and to avoid drugs (unspecified) with mitochondrial toxicity in a patient with a MT-ATP6 pathogenic variant causing DD and epilepsy.39
Alterations to a patient’s existing diet were mentioned in nine studies.8,23,26,27,30,31,33,40,41 In a case series of 43 French patients with dysmorphic features and neurodevelopmental disorders including severe to profound ID, a patient diagnosed with compound heterozygous pathogenic variants in ADCK3 was diagnosed with coenzyme Q10 deficiency. Coenzyme Q10 supplementation and a ketogenic diet were introduced following ES results.40 Most studies with a demonstrated change of patient management reported a change in medication and/or dietary management; however, difference in mortality or morbidity following such a change in management was not reported.
Change of procedures or surveillance
Changes to planned procedures (surgery, imaging, and/or diagnostic studies) or surveillance strategies were specified in 19 studies.7,8,11,20,22,23,24,26,27,28,32,33,35,37,38,41,42,43,44 For example, following ES, 54% (84/155) of patients with CA/DD/ID underwent additional (unspecified) molecular testing, imaging, or biochemical testing.24 In other studies, the ES/GS results led to discontinuation of unnecessary procedures. In a prospective case series of 80 infants with multiple CA and dysmorphic features from Australia, ES resulted in a decision to change surveillance for three patients.7 In one case, echocardiography surveillance for suspected infantile Marfan syndrome was discontinued since the diagnosis was excluded due to the finding of a pathogenic variant in CHRDL1 causing X-linked megalocornea (OMIM 309300). The need for diagnostic tissue biopsy was eliminated for two patients diagnosed with mitochondrial disorders through ES, including combined oxidative phosphorylation deficiency 11 (OMIM 614922) and mitochondrial short-chain enoyl-CoA hydratase 1 deficiency (OMIM 616277)43 and for an additional three patients with CA/DD/ID in the Illumina iHope program10 and for three patients with CA who received rapid ES results in a case series from Australia.23 In a case series of patients with CA/DD/ID, additional invasive testing/procedures were halted in 2% (2/115) of patients after ES. One patient stopped invasive testing for pulmonary causes of respiratory insufficiency after ES identified a pathogenic variant in KIAA1279 causing Goldberg–Shprintzen megacolon syndrome (OMIM 609460), and another patient stopped testing for a mitochondrial disorder after ES identified a pathogenic variant in SNAP25.26 In another case series of patients with CA, rapid GS yielded a diagnosis of ABCC8-associated familial hyperinsulinism type 1 (OMIM 256450) in a patient with hyperinsulinemia.22 The authors reported that the GS results and clinical presentation suggested the focal form of the disorder. Additional imaging was performed that confirmed focal pancreatic lesions and the patient underwent targeted resection, which avoided development of insulin-dependent diabetes, and resulted in a shorter length of stay in the hospital. The patient remained euglycemic more than one year later.22 Overall, many studies reported a change in planned procedures or surveillance strategies, mainly due to a change in the patient’s diagnosis; however, rarely did the authors present data or a description of any resultant reduction in mortality or morbidity.
Referral to specialists
As a consequence of ES/GS results, patients were referred to specialists in a variety of disciplines for follow-up care. This was reported in six studies.8,11,23,32,41,45 In the NSIGHT randomized controlled trial comparing rapid GS to standard genetic testing in critically ill infants with CA and other disorders at a regional NICU and PICU, 27% (4/15) of patients who underwent rapid GS or who were allowed to crossover to GS had a change in subspecialty consult (unspecified) after receipt of GS results.41 In a case series of 53 patients with CA and neurodevelopmental symptoms, 25% (6/24) of patients who received a diagnosis from ES had referrals to a variety of subspecialists, including nephrology, cardiology, ophthalmology, hematology, audiology, and pulmonology.32 In a case series of 278 infants from intensive care units, initiation of specialist care occurred for 26% (27/102) of patients receiving a diagnosis from ES.8 Of the 41 patients in the Illumina iHope program who received a diagnosis from GS, 20% (8/41) were given referrals to specialists to assess for comorbidities, including neurology, ophthalmology, and audiology.11 Taken together, subspecialty referrals were initiated or changed for ~25% of patients who underwent ES/GS and received a diagnosis.
Redirection of care
Nine studies reported a withdrawal of care or start of palliative care for patients after ES/GS results.8,11,14,20,22,23,45,46,47 In a case series of patients with CA/DD/ID, care was withdrawn at the family’s request for a patient after GS resulted in a diagnosis of infantile mitochondrial cardiomyopathy with lactic acidosis.45 Among a series of 66 neonatal patients with CA/DD/ID, dysmorphic features, and other clinical symptoms from a clinical laboratory, rapid ES identified a pathogenic variant in SOX10, which is associated with peripheral demyelinating neuropathy, central dysmyelination, or Waardenburg syndrome with or without Hirschsprung disease. In light of the prognosis and expected outcomes for the patient, the family requested ventilator support be withdrawn.47 In a case series of 23 patients with CA and severe neurological defects from the Netherlands, 71% (5/7) had unsuccessful intensive care treatment stopped after receipt of GS results that informed a diagnosis. However, no details about the treatments or the consequences resulting from their cessation were provided.46
In a case series of 35 consecutive patients with CA/DD/ID who were enrolled in a research study assessing rapid GS from a level 4 NICU or PICU at a quaternary children’s hospital in the United States, of the 21 patients who received a diagnosis with GS, 29% (6/21) began palliative care after receiving the diagnosis. Notably, none of the patients without a genetic diagnosis (0 of 14 patients) transitioned to palliative care.22 In another case series, a patient with multiple CA received a diagnosis of Coffin–Siris syndrome following rapid GS and was transitioned to comfort care and subsequently expired.20 In a case series of patients with CA/DD/ID, a patient who received a diagnosis of neuronal ceroid lipofuscinosis following GS was transitioned to palliative care.11 Overall, withdrawal of care and/or initiation of palliative care following ES/GS was observed in several studies of gravely ill patients.
Clinical trials
Outcomes pertaining to enrollment in or eligibility for clinical trials was specifically reported by six studies.23,24,26,28,32,35 In a large case series of patients with CA by Baldridge et al., 31% (36/115) were enrolled in research studies following ES.24 In a smaller case series of 29 patients in France with CA/DD/ID, two patients (7%) were referred to clinical trials after receiving a diagnosis by ES, including one with a pathogenic variant in the NGLY1 gene (congenital disorder of glycosylation, type Ia; OMIM 212065) and another patient with a pathogenic variant in the RNASEH2B gene (Aicardi–Goutieres syndrome 2; OMIM 610181).35 In a case series of 115 patients with CA/DD/ID, ES results prompted physicians to provide information about clinical trial eligibility for a patient with autosomal recessive, early-onset retinitis pigmentosa.26 Though reported by few studies overall, eligibility for and enrollment in clinical trials may present opportunities for potential benefit for patients who have no treatment options.
Family-focused outcomes
Twelve studies reported outcomes following ES/GS that had an impact on family members of the patient, such as cascade genetic testing, referral to specialists, or changes in clinical management resulting from the diagnosis of a previously unknown disorder.7,11,16,19,26,27,31,32,34,41,42,48 In a cohort of 62 families with congenital anomalies of kidney and urinary tract (i.e., CAKUT) who had ES, first-degree relatives of 1 patient were referred for ophthalmological assessment in light of a diagnosis of renal coloboma. The authors reported optic nerve abnormalities were identified in relatives who were PAX2 variant carriers.34 In a case series of 119 patients with DD/ID and neurodevelopmental disorders, a cardiology referral for evaluation of a congenital heart defect was recommended for a patient’s mother after the patient was diagnosed with Renpenning syndrome (OMIM 309500) caused by a pathogenic variant in PQBP1 gene.27 Cascade testing enabled a diagnosis in 12 relatives of the infants in an Australian cohort with multiple CA who had ES results.7 Although the main impact of ES/GS is for the clinical care of the patient, several studies reported impacts to at-risk family members resulting from a diagnosis in the patient. However, subsequent reductions to mortality or morbidity resulting from the identification of these family members and changes in their clinical care were not reported.
Reproductive-focused outcomes
We identified an impact of ES/GS on outcomes related to reproductive planning for families of patients in 20 studies, including the decisions to become pregnant, terminate a pregnancy, use assisted reproductive technologies, use preimplantation genetic diagnosis, use donor sperm/egg, and undergo previously unplanned additional prenatal testing such as chorionic villus sampling (CVS) or amniocentesis.7,8,11,14,16,22,26,28,31,32,35,36,37,40,41,42,43,44,46,48 ES and GS influenced the decision about having additional children in two studies.37,46 In a case series from the United Kingdom, 1 of 40 parents were considering not having additional children after the patient underwent ES;37 while in a case series from the Netherlands, 9% (2/23) families changed their minds about not having additional children, after learning about the availability of prenatal genetic testing.46
For 95% (20/21) of families who received a result from ES in a German study of 72 patients with CA/DD/ID, the results were important for family planning for either the parents or for the unaffected siblings of patients, and 19% (4/21) of families decided to undergo additional prenatal testing for a subsequent pregnancy based on the patients’ ES results.36 In a case series of patients with CA/DD/ID, Bourchany et al. determined ES enabled reproductive planning in 45% (13/29), and two documented instances of families using ES results to inform prenatal diagnosis in subsequent pregnancies, which confirmed an unaffected fetus in each family.35 In a large case series of patients with CA/DD/ID from the Middle East, North Africa, and Central Asia, 9% (10/118) of families had genetic counseling, carrier testing, or prenatal diagnostic options following ES results in the proband.16 In another case series of 115 patients with CA/DD/ID from the United States who had ES, three families decided to undergo prenatal genetic diagnosis and one family underwent CVS in subsequent pregnancies following receipt of ES results.26 In an Australian study, a mother of a patient with CA/DD/ID who had a CHRND gene variant underwent prenatal genetic diagnosis in a subsequent pregnancy and terminated the pregnancy when the result showed that the fetus was affected.45 Overall, more than half of larger included studies reported an impact of ES/GS relating to the reproductive planning or decisions of patients’ families, expanding outcomes resulting from ES/GS beyond the patient.
Behavioral/psychosocial outcomes
No included larger studies (N ≥ 20) specifically addressed the impact of ES/GS on behavior and/or psychosocial outcomes for patients or their families, such as depression, anxiety, or changes in interpersonal communication. One study reported a single patient’s family was referred to a support group following ES.32 A case report by Nemirovsky et al. mentioned a family’s anxiety was reduced following the diagnosis of a pathogenic variant in the SHANK3 gene by GS.49 Although obtaining a genetic diagnosis by ES/GS may have personal utility, we did not find evidence that ES/GS influenced behavioral or psychosocial outcomes for CA/DD/ID patients or for their families.
Harms of ES, GS
We defined harms of ES and GS as insurance discrimination; a negative impact on family dynamics or communication; a financial burden of the costs of ES or GS; a financial burden of the costs associated with additional testing, surveillance, medication, or dietary modifications stemming from the results of ES or GS; general negative psychosocial impact to the patient or their family; and reduction or loss of privacy. We identified three studies with n ≥ 20 patients and two studies with n < 20 describing the harms associated with ES and/or GS. Baldridge et al. reported two cases of nonpaternity in their case series of 155 patients that required ethics consultation and altered strategies for pretest counseling.24 Nonpaternity was identified in a single case by van Diemen and colleagues. In that patient, it was necessary to disclose nonpaternity to the family to confirm the diagnosis.46 In a case report by He and colleagues, subsequent to ES, parents declined a potentially therapeutic hematopoietic stem cell transplant for economic reasons.50
DISCUSSION
ES/GS are increasingly employed in the clinical evaluation of patients with a variety of conditions, including CA/DD/ID, as evidenced by the increasing publication rates over the past several years. Our findings demonstrate that ES/GS can influence outcomes for individuals with CA/DD/ID and their family members. Overall, included studies documented a change in clinical management as a result of ES/GS, including change in medications, procedures, or referral to specialists. When considering the types of medical management decisions, more than half of patients experienced a reported clinical impact related to the ES/GS diagnosis. Likewise, more than half of larger included studies reported an impact of ES/GS relating to the reproductive planning or decisions of patients’ families, further expanding the usefulness of ES/GS beyond the patient. However, few studies describe beneficial health outcomes or improved quality of life resulting from ES/GS for patients with CA/DD/ID. Nonetheless, despite little direct evidence that ES/GS improved mortality or ameliorated morbidity, the studies included in this review provide indirect evidence of the clinical and personal utility of ES/GS for patients with CA/DD/ID and their family members.
Though there are many publications describing ES/GS diagnostic yield, we identified relatively few studies documenting outcomes resulting from ES/GS for CA/DD/ID patients or their families. There are several possible reasons for this. First, we explicitly excluded studies that reported only diagnostic yield, as diagnostic yield is already well documented in the literature. Our goal was to assess outcomes resulting from a genetic diagnosis, such as improvement in morbidity or mortality, changes in surveillance or prevention, decisions about medical or surgical interventions, subspecialty referrals, better prognostication, and reproductive counseling. Furthermore, our search did not necessarily encompass all possible measures of utility and thus, this review does not preclude other forms of utility that were not included in the scope of the search strategy. Second, we excluded studies of patients with CA in which the reported age of onset was unstated or diagnosed when greater than 1 year of age. We focused our analysis on studies of patients who present with CA early in life, at a time when a diagnosis made by ES/GS may have the greatest impact for the patient by ending the diagnostic odyssey and for family by providing opportunities for reproductive options in subsequent pregnancies.7,8,14,31 However, patients who present after 1 year of age may also derive benefit from a diagnosis made through ES/GS.
We also found substantial inherent limitations to the included studies. First, most were case reports or small case series where the goal was to make a genetic diagnosis for CA/DD/ID and the reported outcomes of interest were secondary. These study designs are problematic, as they introduce risk of bias and lack of generalizability. However, randomized controlled trials (RCTs), which are considered to generate the highest levels of evidence, are unlikely to be performed in this patient population. There was only one RCT (NSIGHT1) that assessed the use of rapid GS, and that study assessed only the rate of diagnosis, and was terminated prematurely due to loss of equipoise and difficulty adhering to randomization to the control group.41 Therefore, observational studies, including case reports and small case series, are important, and often the only form of evidence for patients with rare diseases. Second, among the included studies, there was substantial heterogeneity of patient phenotypes and indications for ES/GS. This translated to very small numbers of patients eligible for similar interventions, hindering robust measurement of the outcomes of interest. Moreover, aside from halting medical management or entering palliative care, outcome measures of morbidity, mortality, or family planning have long time horizons that are difficult to measure. Consequently, the body of evidence supporting or refuting the usefulness of ES/GS for patients with CA/DD/ID is significantly heterogeneous in terms of quality, methods, and reporting, similar to the case for CMA.3
We found that reporting of outcomes we assume are of value to key stakeholders using ES/GS was not uniform or complete.51 For example, we found only two studies demonstrating a reduction in mortality or morbidity resulting from ES/GS. Few studies reported whether the clinical course was improved by medication or diet, or by enrolling in a clinical trial. When a change in diagnostic thinking or clinical management of the patient follows ES/GS, to demonstrate the usefulness and value of the testing, investigators should study the corresponding impact on primary health outcomes where possible, either through comparisons to historical cases or matched controls. Moreover, with the near ubiquity of electronic medical records, this information may be readily available to provide further evidence of the utility of ES/GS. Further, impacts of ES/GS on reproductive-related decisions were generally not well described; many studies only reported vague descriptions or general statements about genetic counseling. We suspect use of ES/GS results informs important family planning decisions more commonly than was reflected in the evidence reviewed. Reproductive decision-making is a valued outcome of genetic testing, and as such, should be systematically measured as an outcome of ES/GS intervention studies.51,52
Future studies of ES/GS should document the impact of test results more routinely and uniformly, and in so doing, make available a more robust body of evidence to support utilization of this critically important technology. To accomplish this, a uniform framework is needed to allow prospective data collection to be standardized and thus, more easily aggregated and compared in meta-analyses. The genetics community should develop a standard set of outcomes for ES/GS, along with corresponding metrics to assess those outcomes to facilitate evidence development. The issue of “what to measure” is somewhat unique for the heterogeneous groups of patients with rare disorders. Defining the outcome measures will allow use of well-established reporting frameworks for studies of ES/GS in rare disease populations, like the PRISMA framework for systematic evidence reviews53 and the CHEERS framework used for economic evaluations.54 Use of frameworks informing data collection and reporting could simultaneously improve the quality of the literature, as most studies we identified were of case reports or small case series with limited assessments when considered specifically in the context of health and clinical outcomes reporting. A recently published scoping review on the use of ES/GS for pediatric patients with phenotypes very similar to the articles we reviewed identified similar limitations within the current body of evidence and appealed for the development of a standard framework for clinical impact data reporting in sequencing studies.12
Conclusion
In summary, we performed a systematic evidence review to characterize the impact of ES/GS in patients with CA/DD/ID on clinical management and health outcomes. We identified one RCT and numerous case series and case reports describing clinical and patient outcomes, for which the overall evidence was limited. However, a change in patient management was observed in nearly all included studies (including case reports), and a substantial number of publications reported a clinical impact on the patient’s family members or an impact on reproductive outcomes. Future studies of ES/GS results for patients with CA/DD/ID should explicitly measure patient and family outcomes resulting from testing to better assess the clinical and personal utility of ES/GS.
References
Manning M, Hudgins L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12:742–745.
Hochstenbach R, van Binsbergen E, Engelen J, et al. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur J Med Genet. 2009;52:161–169.
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764.
Fan LL, Huang H, Jin JY, et al. Whole exome sequencing identifies a novel mutation (c.333+2T>C) of TNNI3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease. Gene. 2018;648:63–67.
Cheng SSW, Chan KYK, Leung KKP, et al. Experience of chromosomal microarray applied in prenatal and postnatal settings in Hong Kong. Am J Med Genet C Semin Med Genet. 2019;181:196–207.
Jang W, Kim Y, Han E, et al. Chromosomal microarray analysis as a first-tier clinical diagnostic test in patients with developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies: a prospective multicenter study in Korea. Ann Lab Med. 2019;39:299–310.
Stark Z, Tan TY, Chong B, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18:1090–1096.
Meng L, Pammi M, Saronwala A, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 2017;171:e173438.
Clark MM, Stark Z, Farnaes L, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med. 2018;3:16.
Nambot S, Thevenon J, Kuentz P, et al. Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet Med. 2018;20:645–654.
Scocchia A, Wigby KM, Masser-Frye D, et al. Clinical whole genome sequencing as a first-tier test at a resource-limited dysmorphology clinic in Mexico. NPJ Genom Med. 2019;4:5.
Smith HS, Swint JM, Lalani SR, et al. Clinical application of genome and exome sequencing as a diagnostic tool for pediatric patients: a scoping review of the literature. Genet Med. 2019;21:3–16.
Jain P, Sharma S, Tripathi M. Diagnosis and management of epileptic encephalopathies in children. Epilepsy Res Treat. 2013;2013:501981.
French CE, Delon I, Dolling H, et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 2019;45:627–636.
Sawyer SL, Hartley T, Dyment DA, et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016;89:275–284.
Dixon-Salazar TJ, Silhavy JL, Udpa N, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4:138ra78.
Anazi S, Maddirevula S, Fageih E, et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry. 2017;22:615–624.
Vissers LELM, van Nimwegen KJM, Schieving JH, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med. 2017;19:1055–1063.
Nair P, Sabbagh S, Mansour H, et al. Contribution of next generation sequencing in pediatric practice in Lebanon. A study on 213 cases. Mol Genet Genomic Med. 2018;6:1041–1052.
Farnaes L, Hildreth A, Sweeney NM, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med. 2018;3:10.
Kaye AJ, Rand EB, Munoz PS, Spinner NB, Flake AW, Kamath BM. Effect of Kasai procedure on hepatic outcome in Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;51:319–321.
Willig LK, Petrikin JE, Smith LD, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3:377–387.
Stark Z, Lunke S, Brett GR, et al. Meeting the challenges of implementing rapid genomic testing in acute pediatric care. Genet Med. 2018;20:1554–1563.
Baldridge D, Heeley J, Vineyard M, et al. The exome clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet Med. 2017;19:1040–1048.
Tammimies K, Marshall CR, Walker S, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 2015;314:895–903.
Iglesias A, Anyane-Yeboa K, Wynn J, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med. 2014;16:922–931.
Soden SE, Saunders GJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168.
Srivastava S, Cohen JS, Vernon H, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol. 2014;76:473–483.
Petrikin JE, Willig LK, Smith LD, Kingsmore SF. Rapid whole genome sequencing and precision neonatology. Semin Perinatol. 2015;39:623–631.
Zhu X, Petrovski S, Xie P, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17:774–781.
Kuperberg M, Lev D, Blumkin L, et al. Utility of whole exome sequencing for genetic diagnosis of previously undiagnosed pediatric neurology patients. J Child Neurol. 2016;31:1534–1539.
Nolan D, Carlson M. Whole exome sequencing in pediatric neurology patients: clinical implications and estimated cost analysis. J Child Neurol. 2016;31:887–894.
Tarailo-Graovac M, Shyr C, Ross CJ, et al. Exome sequencing and the management of neurometabolic disorders. N Engl J Med. 2016;374:2246–2255.
Bekheirnia MR, Bekheirnia N, Bainbridge MN, et al. Whole-exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genet Med. 2017;19:412–420.
Bourchany A, Thauvin-Robinet C, Lehalle D, et al. Reducing diagnostic turnaround times of exome sequencing for families requiring timely diagnoses. Eur J Med Genet. 2017;60:595–604.
Evers C, Staufner C, Graznow M, et al. Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Mol Genet Metab. 2017;121:297–307.
Miller KA, Twigg SR, McGowan SJ, et al. Diagnostic value of exome and whole genome sequencing in craniosynostosis. J Med Genet. 2017;54:260–268.
Perucca P, Scheffer IE, Harvey AS, et al. Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 2017;131:1–8.
Cordoba M, Rodriguez-Quiroga SA, Vega PA, et al. Whole exome sequencing in neurogenetic odysseys: An effective, cost- and time-saving diagnostic approach. PLoS One. 2018;13:e0191228.
Thevenon J, Duffourd Y, Masurel-Paulet A, et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet. 2016;89:700–707.
Petrikin JE, Cakici JA, Clark MM, et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom Med. 2018;3:6.
Todd EJ, Yau KS, Ong R, et al. Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet J Rare Dis. 2015;10:148.
Bick D, Fraser PC, Gutzeit MF, et al. Successful application of whole genome sequencing in a medical genetics clinic. J Pediatr Genet. 2017;6:61–76.
Tan TY, Dillon OJ, Stark Z, et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 2017;171:855–862.
Thiffault I, Farrow E, Zellmer L, et al. Clinical genome sequencing in an unbiased pediatric cohort. Genet Med. 2019;21:303–310.
van Diemen CC, Kerstjens-Frederikse WS, Bergman KA, et al. Rapid targeted genomics in critically ill newborns. Pediatrics. 2017;140:e20162854.
Powis Z, Farwell Hagman KD, Speare V, et al. Exome sequencing in neonates: diagnostic rates, characteristics, and time to diagnosis. Genet Med. 2018;20:1468–1471.
Valencia CA, Husami A, Holle J, et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front Pediatr. 2015;3:67.
Nemirovsky SI, Cordoba M, Zaiat JJ, et al. Whole genome sequencing reveals a de novo SHANK3 mutation in familial autism spectrum disorder. PLoS ONE. 2015;10:e0116358.
He X, Zou R, Zhang B, You Y, Yang Y, Tian X. Whole Wiskott-Aldrich syndrome protein gene deletion identified by high throughput sequencing. Mol Med Rep. 2017;16:6526–6531.
Scheuner MT, Russell MM, Chanfreau-Coffinier C, et al. Stakeholders’ views on the value of outcomes from clinical genetic and genomic interventions. Genet Med. 2019;21:1371–1380.
Kohler JN, Turbitt E, Lewis KL, et al. Defining personal utility in genomics: a Delphi study. Clin Genet. 2017;92:290–297.
Shamseer L, Moher D, Clarke M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ. 2015;350:g7647.
Husereau D, Drummond M, Petrou S, et al. Consolidated Health Economic Evaluation Reporting Standards (CHEERS) statement. BMJ. 2013;346:f1049.
Author information
Authors and Affiliations
Consortia
Ethics declarations
Disclosure
S.E.H. received research support from Alexion Pharmaceuticals to serve as a site investigator for a prospective registry for hypophosphatasia. D.T.M. received honoraria from Prevention Genetics and Takeda Pharmaceuticals. M.C.S. receives salary (no equity) as a lab director at Washington University School of Medicine, received salary (no equity) as a lab director at HudsonAlpha Institute of Biotechnology (performs clinical whole genome sequencing), and received consulting fees from PierianDx. J.S. received salary from the Partners Healthcare Laboratory for Molecular Medicine and from the Brigham and Women’s Hospital Cytogenomics Laboratory. The other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Disclaimer:
The ACMG has recruited expert panels, chosen for their scientific and clinical expertise, to conduct systematic evidence reviews (SERs) to support the development of clinical practice guidelines. An SER focuses on a specific scientific question and then identifies, analyzes and summarizes the findings of relevant studies. ACMG SERs are provided primarily as educational resources for medical geneticists and other clinicians to help them provide quality medical services. They should not be considered inclusive of all relevant information on the topic reviewed.
Reliance on this SER is completely voluntary and does not necessarily assure a successful medical outcome. In determining the propriety of any specific procedure or test, the clinician should apply his or her own professional judgment to the specific clinical circumstances presented by the individual patient or specimen. Clinicians are encouraged to document the reasons for the use of a particular procedure or test, whether or not it is in conformance with this SER. Clinicians also are advised to take notice of the date this SER was published, and to consider other medical and scientific information that becomes available after that date.
Correspondence: ACMG (documents@acmg.net)
Rights and permissions
About this article

Cite this article
Malinowski, J., Miller, D.T., Demmer, L. et al. Systematic evidence-based review: outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genet Med 22, 986–1004 (2020). https://doi.org/10.1038/s41436-020-0771-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0771-z
Keywords
This article is cited by
-
Evidence review and considerations for use of first line genome sequencing to diagnose rare genetic disorders
npj Genomic Medicine (2024)
-
Decoding complex inherited phenotypes in rare disorders: the DECIPHERD initiative for rare undiagnosed diseases in Chile
European Journal of Human Genetics (2024)
-
Fetal alcohol spectrum disorders
Nature Reviews Disease Primers (2023)
-
A review of key terminology and definitions used for birth defects globally
Journal of Community Genetics (2023)
-
De novo variants underlying monogenic syndromes with intellectual disability in a neurodevelopmental cohort from India
European Journal of Human Genetics (2023)
