
Abstract
Purpose
MED12 is a subunit of the Mediator multiprotein complex with a central role in RNA polymerase II transcription and regulation of cell growth, development, and differentiation. This might underlie the variable phenotypes in males carrying missense variants in MED12, including X-linked recessive Ohdo, Lujan, and FG syndromes.
Methods
By international matchmaking we assembled variant and clinical data on 18 females presenting with variable neurodevelopmental disorders (NDDs) and harboring de novo variants in MED12.
Results
Five nonsense variants clustered in the C-terminal region, two splice variants were found in the same exon 8 splice acceptor site, and 11 missense variants were distributed over the gene/protein. Protein truncating variants were associated with a severe, syndromic phenotype consisting of intellectual disability (ID), facial dysmorphism, short stature, skeletal abnormalities, feeding difficulties, and variable other abnormalities. De novo missense variants were associated with a less specific, but homogeneous phenotype including severe ID, autistic features, limited speech and variable other anomalies, overlapping both with females with truncating variants as well as males with missense variants.
Conclusion
We establish de novo truncating variants in MED12 as causative for a distinct NDD and de novo missense variants as causative for a severe, less specific NDD in females.
Similar content being viewed by others

INTRODUCTION
MED12 is 1 of 31 subunits (MED1-MED31) of the large multiprotein Mediator complex that regulates gene expression in all eukaryotes by interacting with RNA polymerase II.1,2 It is involved in transcriptional elongation and termination, messenger RNA (mRNA) processing, noncoding RNA activation, super enhancer formation, and epigenetic regulation.2,3 Hence, Mediator complex is a master coordinator of cell growth and homeostasis, development, and differentiation,4 and its subunit MED12 is a critical transducer of regulatory information essential for organogenesis.5 MED12 consists of the Med12 domain that forms part of the kinase section of Mediator, which can negatively regulate the Gli3-dependent sonic hedgehog signaling pathway via its interaction with Gli3 within RNA polymerase II;6 the Med12-LCEWAV domain, of which the function is not known; and the Med12-PQL domain that binds to β-catenin, which targets MED12 to activate transcription of Wnt-responsive genes7.
Hemizygous missense variants in MED12 are known to cause at least three different but overlapping X-linked syndromic neurodevelopmental disorders (NDDs) in males: FG syndrome (FGS1, also known as Opitz–Kaveggia syndrome [OKS; OMIM 305450]), Lujan–Fryns syndrome (LS; OMIM 309520), and X-linked Ohdo syndrome (OHDOX; OMIM 300895). FGS1 is characterized by intellectual disability (ID); hypotonia; dysmorphic features such as macrocephaly, prominent forehead, downslanting palpebral fissures, small ears with occasionally sensorineural hearing loss, broad thumbs, and halluces; constipation and/or anal anomalies; partial agenesis of the corpus callosum; and characteristic hyperactive and talkative behavior.8,9 LS is characterized by mild to moderate ID; dysmorphic features such as macrocephaly, large head, a long thin face, low-set ears, prominent nasal bridge, high narrow palate, short and deep philtrum; tall stature with thin or marfanoid habitus, behavioral aspects and abnormalities of the corpus callosum.10,11 OHDOX is characterized by ID and typical facial features, including blepharophimosis, facial coarsening, thick alae nasi, and a triangular face.12,13 Additionally, variants in MED12 have been implicated in nonspecific NDDs in males.11,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28 Though heterozygous females in these families are usually not affected, three females with inherited MED12 variants,15,17,25 p.(Ser1967Glnfs*84), p.(Arg1148His), and p.(Ile771Thr), and three females with de novo MED12 variants,29,30,31 p.(Glu172Gln), c.1249–1G>C and p.(Arg521His), were reported to have NDDs with variable cognitive impairment.
Here, we report on assembled clinical and genetic data of 18 females with de novo variants in MED12. Truncating variants in seven individuals resulted in a syndromic presentation with variable ID, short stature, facial dysmorphism, feeding difficulties and skeletal abnormalities. Missense variants in 11 individuals were associated with a phenotype of severe ID, autistic features and limited speech, and variable dysmorphic features.
MATERIALS AND METHODS
DNA analysis and X-inactivation
Affected individuals were referred by physicians working in disability services and clinical geneticists, and connected through personal communication, Decipher database,32 and GeneMatcher.33 MED12 variants were identified by exome or genome sequencing and confirmed by Sanger sequencing (Supplementary Table 1). X-chromosome inactivation (XCI) analysis was performed at the androgen receptor locus with a modification of the assay previously described.34 Primer sequences and polymerase chain reaction (PCR) conditions for Sanger sequencing and XCI testing are available upon request.
RNA isolation, cDNA synthesis, and RT-PCR
All RNA isolations were performed with the NucleoSpin RNA Clean-up Kit (catalog number 740955–50, Macherey-Nagel, Düren, Germany) according to manufacturer’s protocol. RNA was quantified by nanodrop. One microgram of total RNA was used for all complementary DNA (cDNA) synthesis reactions. For reverse transcription PCR (RT-PCR) analysis, cDNA was synthesized by the SuperScript VILO Master Mix (catalog number 11755050, Thermo Fisher Scientific, Waltham, MA, USA), following manufacturer’s instructions.
To determine the potential splice acceptor variant effect on MED12 splicing in individuals 6 and 7, RT-PCR analysis was performed using primers located in exon 8 (forward) and exon 12 (reverse) of MED12. Primer sequences are available upon request. For RT-PCR analysis, 25 ng of cDNA was used for all reactions. Amplifications were performed according to standard procedures. PCR products were sequenced using the ABI PRISM BigDye Terminator Cycle Sequencing V2.0 Ready Reaction Kit and analyzed with the ABI PRISM 3730 DNA analyzer (Applied Biosystems, Foster City, CA, USA).
Minigene-based splice assay
The effect of the two splice acceptor variants was also assessed by minigene-based splicing assays employing wild-type and mutant constructs using DNA from individuals 6 and 7 as a template. To amplify the genomic region encompassing exons 8 and 9 of the MED12 gene, we designed PCR primers located in exons 7 (forward) and 11 (reverse) using Primer3plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). The PCR reaction mixtures of 50 μl contained 25 μM of each primer pair, a high-fidelity Taq polymerase Phusion® High-Fidelity DNA Polymerase (catalog number M0530L, New England BioLabs, Ipswich, MA, USA), 5× PCR HF buffer, 1 μl of dNTP mix, 5× Q solution and 5 ng genomic DNA from patients 1 and 2. Amplifications were performed using a two-phase touchdown PCR program described before. The resulting PCR products were run on a 0.8% agarose gel and were purified with the NucleoSpin Gel and PCR cleanup kit (catalog number 740609.250; Macherey-Nagel, Düren, Germany), according to the manufacturer’s protocol. One hundred fifty nanograms of each purified insert were cloned into the pDONR201 vector by using the Gateway BP Clonase Enzyme Mix (catalog number 11789021; Thermo Fisher Scientific).
The different pDONR cloned constructs were validated by Sanger sequencing, and 150 ng of each were cloned into the destination vector pCI-NEO-RHO exon3,5/DEST by using the Gateway LR Clonase enzyme mix (catalog number 11791043; Thermo Fisher Scientific). Three minigene constructs were generated, one with the wild-type and two with the c.1249–1G>C and c.1249–2A>G variants. HEK293T cells (ATCC CRL-3216; American Type Culture Collection [ATCC], Wesel, Germany) were grown in DMEM supplemented with 10% (volume/volume) fetal calf serum (catalog number F7524; Sigma Aldrich, St. Louis, MO, USA), 1% (volume/volume) sodium pyruvate (catalog number S8636; Sigma Aldrich), and penicillin/streptomycin (catalog number P4333; Sigma Aldrich). Cells were seeded in 6-well plates at a density of 5×105 cells/well and transfected the day after with 2 μg of each minigene using FuGENE HD Transfection Reagent (catalog number E2311; Promega, Madison, WI, USA) with a DNA:liposome ratio of 1 μg:3 μl, according to the manufacturer’s instructions. Medium was replaced after 24 hours, and cells were harvested 48 hours after transfection. Total RNA isolation, cDNA production, and RT-PCR reactions were performed as described previously. The RT-PCR products were resolved on a 2% (weight/volume) agarose gel, the resulting bands were excised, and the nucleic acid was purified by using NucleoSpin Gel & PCR cleanup (catalog number 740609.50, Macherey-Nagel, Düren, Germany). Sanger sequencing was performed on 100 ng of each purified band by using the same primers used in the PCR reaction showing no other variant except those introduced by us.
RESULTS
Variant spectrum, splicing effects, and X-inactivation
Five individuals carried de novo nonsense variants in the last four exons and clustering in or downstream of the catenin-binding domain of MED12 (NM_005120.2) (Fig. 1). In two individuals, de novo variants affecting the splice acceptor of exon 8 were identified: c.1249–1G>C and c.1249–2A>G. Additionally, 11 females carried de novo missense variants predicted to be deleterious (Supplementary Table 1). The identified missense variants were distributed over the N-terminal two-thirds of the protein, and all but one were located between the LCEWAV and the catenin-binding domain (Fig. 1). p.(Arg1138Trp) recurrently occurred in two individuals. None of the identified variants were present in the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org; Supplementary Table 1). The MED12 probability of loss of function intolerance (pLi) score was 1 and the Z-score was 6.58, which indicates that there is strong selection against protein truncating variants and missense variants in males and females.

Known intellectual disability (ID)‐associated missense variants in males are displayed below the protein scheme. Above are previously reported inherited frameshift,15 inherited17,25 and de novo29,30 missense, and de novo splice site31 variants in female individuals, together with the de novo variants in MED12 (NM_005120.2) identified in this study (in bold). MED12 domains; Med12: Transcription mediator complex subunit Med12. This subunit forms part of the kinase section of Mediator, which can negatively regulate the Gli3-dependent sonic hedgehog signaling pathway via its interaction with Gli3 within the RNA polymerase II. A complex is formed between Med12, Med13, CDK8, and CycC, which is responsible for suppression of transcription.6 Med12-LCEWAV; Eukaryotic Mediator 12 subunit domain: the function of this region is not known, but there is a conserved sequence motif (LCEWAV). Med12-PQL; Eukaryotic Mediator 12 catenin-binding domain: β-catenin physically and functionally targets this PQL (proline-, glutamine-, leucine-rich) region of the Med12 subunit of Mediator to activate transcription of Wnt-responsive genes7.
We used RT-PCR to investigate whether the two variants in the canonical splice acceptor of exon 8 affected splicing. We found expression of only the wild-type fragment in EBV-LCL and fibroblast lines from individual 1 carrying the c.1249–1G>C variant, and in a fibroblast line from individual 2 who carried the c.1249–2A>G variant (Fig. 2b, c). To investigate the splicing defect in more detail, we generated two MED12 minigenes spanning exons 7 to 11 using control and patient genomic DNAs. HEK293T cells were transfected with mutant and control minigenes (Fig. 2d). While the control minigene did not show any splicing defect, both mutated minigenes showed complete skipping of exon 9 (Fig. 2e), leading to an abnormal transcript due to an early termination codon: p.(Val417Alafs*76). This indicates that the presence of only the wild-type allele in fibroblasts and EBV-LCLs might be due to either nonsense-mediated mRNA decay or more likely skewing of XCI.

(a) Position of the variants (red circle) at the donor site of MED12 exon 9. Blue arrows represent primers used for analysis. (b,c) No effect on splicing is found on reverse transcription polymerase chain reaction (RT-PCR) analysis of expression of MED12 in EBV-LCLs and fibroblast cell lines from patients and controls. One microgram cycloheximide (CHX) per milliliter medium was added for four hours to inhibit nonsense-mediated messenger RNA (mRNA) decay. (d,e) Minigene analysis shows that the canonical splice site variants induce skipping of exon 9 in the transcript containing the variant. This causes an open reading frame (ORF) change and the generation of a premature stop codon, p.(Val417Alafs*76). A nontransfected HEK293T cell line was used as control.
Analysis of the X-inactivation status showed extreme skewing (>95:5) in blood or fibroblasts of three individuals with protein truncating variants including individual 2, and random XCI in three individuals (Supplementary Table 1). In individual 1, the two androgen receptor alleles were noninformative. XCI was extremely skewed (>95%) in seven of the 11 individuals with missense variants and was skewed (85%) in one. In three individuals with a missense variant, XCI was random in peripheral blood cells. In four individuals where two different tissues—whole blood and either a fibroblast cell line or EBV-LCLs—were tested, XCI behaved comparably between tissues. Three of these individuals had skewing of X-inactivation, and the variant was located on the inactive allele in the tested tissues.
Clinical spectrum
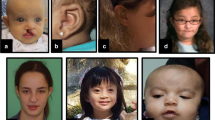
All seven individuals with de novo protein truncating variants in MED12 presented with ID, ranging from mild in one to severe/profound in four individuals (Table 1; Supplementary Table 1). Speech was commonly delayed, and absent in two individuals. Brain imaging by magnetic resonance image (MRI) or ultrasound was performed in five individuals and showed partial hypoplasia or agenesis of the corpus callosum and/or enlarged ventricles in three of them. Feeding difficulties were reported in five individuals, muscular hypotonia in four, autistic features in three, and seizures in only one individual. Birth weight and/or length below −2 SD were observed in two individuals as well as congenital microcephaly. Postnatally, short stature occurred in four of six individuals, of whom these data were available. The two individuals who were of normal length were the youngest of the six. This suggests that short stature is a progressive feature that might become apparent only at a later age. Microcephaly and macrocephaly were each reported once, whereas three individuals had a normal head circumference. Skeletal abnormalities were present in six of the seven individuals and included syndactyly of fingers II–IV or of toes II and III as well as other finger anomalies, scoliosis, or rhizomelic limb shortening. Facial gestalt was quite distinct in all the individuals and included sparse hair; low-set, posteriorly rotated ears; a prominent forehead or bitemporal narrowing; hypertelorism; downslanting palpebral fissures; blepharophimosis; palatal anomalies or a bifid uvula; dental crowding; and micro- or retrognathia (Fig. 3). Variable other features were laryngeal anomalies requiring tracheostoma in two individuals, anteriorly placed anus (4/7), hearing loss (2/3), and pigmentation anomalies (2/3). Individuals 1 and 2 with variants in the splice acceptor of exon 8 had less prominent micro/retrognathia and shared other clinical aspects such as a very prominent forehead with large fontanels, hypertelorism, and MRI anomalies including partial agenesis of corpus callosum, enlarged ventricles, and abnormal tectum and cerebellar vermis.

(a) Females with truncating variants presented with marked facial dysmorphism such as abnormal skull shape, low-set posteriorly rotated ears, blepharophimosis, crowded teeth, retrognathia, and variable other abnormalities such as syndactyly of fingers and/or toes. Individuals 1 and 2 with a splice site variant share a very prominent forehead and magnetic resonance image (MRI) anomalies such as partial agenesis of corpus callosum, enlarged ventricles and tectum, and cerebellar vermis abnormalities. (b) In comparison, females with de novo missense variants presented with minor, but rather nonspecific facial dysmorphisms and less frequently with other structural or morphological abnormalities such as syndactyly and clinodactyly of fingers and/or toes (I12, I11, I16) or brain abnormalities such as agenesis of corpus callosum (I8, I15) or Chiari malformation (I18).
In contrast, the individuals with de novo missense variants in MED12 presented with a less distinct, but still homogeneous phenotype. Eight of 11 individuals presented with severe to profound ID and six of those with absent speech. One exception was individual 14 with a low-normal IQ of 83, but she had other hallmarks of MED12-related disorders, such as thin corpus callosum, a prominent forehead, and ptosis. Behavioral abnormalities such as autistic features or attention deficit were common and reported in eight individuals. Corpus callosum anomalies were reported in four and cortical thinning in one of ten individuals in whom MRI was performed. Growth parameters were variable with mostly normal measurements at birth and postnatally. Short stature was reported in only one individual, while tall stature occurred in two individuals. Syndactyly of fingers or toes was reported in two individuals only, while scoliosis and pectus anomalies were reported in three individuals. Ophthalmological anomalies were found in nine individuals of which strabismus and nystagmus were most frequent. Further variable anomalies in single or few individuals included muscular hypotonia, hearing loss, or constipation. Subtle and nonspecific facial dysmorphisms were noted in several of the individuals (Fig. 3).
DISCUSSION
In this study, we identified 18 unrelated females with de novo variants in MED12 and presenting with variable NDDs. These cases can be classified into two molecularly and phenotypically distinct groups. Truncating variants were identified in seven individuals and resided either in the splice acceptor of exon 8 (n = 2), resulting in out of frame skipping of exon 9, or clustered C-terminally in or downstream of the catenin-binding domain (n = 5). The phenotype of these individuals appeared rather distinct and comprised mainly severe ID; short stature; (severe) feeding difficulties; syndactyly of toes II and III and/or syndactyly of fingers II, III, and IV; and variable other skeletal abnormalities. Marked facial dysmorphism included an abnormal skull shape, sparse hair, low-set posteriorly rotated ears, blepharophimosis, micro- and/or retrognathia, and dental crowding. The presence of abnormal transcripts and subsequently truncated protein might be expected for these seven variants. Particularly for the C-terminal variant p.(Gln2150*), a truncating effect is likely since localization in the last or penultimate exon usually does not lead to nonsense-mediated mRNA decay.35
So far, pathogenic MED12 variants described in the literature have been mainly missense. Only two other truncating variants were reported previously. First, a frameshift variant, c.5898dupC, also located in the C-terminal catenin-binding domain, was identified in ten affected males and an affected female from one family with severe to profound ID and facial dysmorphism.15 Nine heterozygous females showed only borderline to mild ID. This variant has been demonstrated to escape nonsense-mediated mRNA decay and lead to creation of an additional abnormal transcript due to activation of two cryptic splice sites in exon 41.15 Second, a de novo splice variant that is identical to the one identified in individual 1 in this study was reported in a one-year-old female with a similar phenotype comprising developmental delay, facial dysmorphism, cleft lip and palate, and a thin corpus callosum.31 For the two novel splice variants we identified, we could not detect an aberrant transcript. This could be due to the skewed X-inactivation in the peripheral blood samples of these individuals as we could observe a protein truncating transcript in a minigene assay.
Clustering of the truncating variants in two distinct regions of the gene/protein points to specific effects. The very C-terminal variants leave all known functional domains intact, while the N-terminal splice variants in the splice acceptor of exon 8 would be expected to affect the LCEWAV and the Catenin-binding domain. Individuals 1 and 2 with these splice variants share particular clinical aspects such as skull shape and MRI abnormalities. Apart from that, the phenotype between individuals with C-terminal nonsense variants and N-terminal splice variants is highly overlapping. We suspect a common loss-of-function consequence of the truncating variants rather than a dominant-negative effect. This would be in accordance with gnomAD constraint scores, indicating intolerance of MED12 towards loss-of-function variants. As truncating variants in MED12 were mainly observed in females with a severe and syndromic phenotype, their functional consequence might be possibly more severe than the effect of most pathogenic missense variants.
The phenotype in the 11 females with de novo missense variants appeared less specific in comparison, but was still homogeneous with severe or profound ID in all but two individuals, frequent autistic features, and lack or severe limitation of speech. Variable other anomalies included agenesis of corpus callosum, mild short stature, syndactyly, subtle facial dysmorphism, and feeding difficulties. Similarly to previously reported missense variants found in males, the de novo missense variants identified in females in this study were distributed all over the protein. Previously, only two missense variants, p.(Glu172Gln)29 and p.(Arg521His),31 have been reported in females with comparable phenotypes.
Pathogenic variants in MED12 are associated with various nonsyndromic and syndromic NDDs in both males and females. Apart from shared intellectual disability, dysmorphic features such as blepharophimosis, brain anomalies such as agenesis or hypoplasia of corpus callosum, or other morphological anomalies such as syndactyly overlap between both males and females but also within the male and within the female NDDs. In addition to the functional consequences (e.g., truncating versus missense variants) and the location of the variants, XCI patterns in females might also contribute to the variable phenotypic expression. Skewing of XCI in favor of the wild-type allele can accordingly contribute to the compensation of X-chromosomal variants in heterozygous females, thus preventing clinical manifestation. However, XCI patterns in previous reports did not correlate with presence or severity in females heterozygous for MED12 variants.14,15,17,25,29 While XCI skewing was demonstrated more frequently in 12 individuals with either truncating or missense variants, also random XCI was reported in four individuals, who did not present with a less or more severe phenotype. X-inactivation is a random transcriptional silencing process in gastrulation, where one of the two X chromosomes is inactivated. Afterward, this specific pattern is maintained in the daughter cells that all have the same maternal or paternal X inactivated, often resulting in functional mosaicism with the mutant allele being active or inactive depending on the tissue. In blood or fibroblast cells of affected individuals, X-inactivation was skewed, and only the wild-type allele could be detected, which would in principle point to a compensatory effect. Other tissues were not available. We therefore speculate that, in contrast to blood, the mutant allele might be active in progenitor brain cells and that this might underlie the severe phenotype. Pigmentation anomalies can be suggestive for such a functional mosaicism.36 Such skin pigmentation anomalies were observed in at least three individuals in our cohort, and partial skin depigmentation was reported in one of the previously published female individuals.29
In conclusion, we demonstrate that de novo missense and specific truncating variants in MED12 cause NDDs with variable abnormalities and dysmorphic features in females, partly overlapping with the previously known phenotypic spectrum from affected males. We speculate that “severity,” localization, and functional consequences of the variants as well as XCI patterns in females might contribute to the clinical presentation and variability both among females and between affected females and males.
Data availability
All data and materials that are not included in the paper are available upon request.
References
Jeronimo, C. & Robert, F. The mediator complex: at the nexus of RNA polymerase II transcription. Trends. Cell. Biol. 27, 765–783 (2017).
Conaway, R. C. & Conaway, J. W. Function and regulation of the Mediator complex. Curr. Opin. Genet. Dev. 21, 225–230 (2011).
Poss, Z. C., Ebmeier, C. C. & Taatjes, D. J. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 48, 575–608 (2013).
Yin, J. W. & Wang, G. The Mediator complex: a master coordinator of transcription and cell lineage development. Development 141, 977–987 (2014).
Lyons, M. J. MED12-related disorders. GeneReviews. http://www.ncbi.nlm.nih.gov/books/NBK1676/ (2020).
Zhou, H., Kim, S., Ishii, S. & Boyer, T. G. Mediator modulates Gli3-dependent Sonic hedgehog signaling. Mol. Cell. Biol. 26, 8667–8682 (2006).
Kim, S., Xu, X., Hecht, A. & Boyer, T. G. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem. 281, 14066–14075 (2006).
Clark, R. D. et al. FG syndrome, an X-linked multiple congenital anomaly syndrome: the clinical phenotype and an algorithm for diagnostic testing. Genet. Med. 11, 769–775 (2009).
Opitz, J. M. & Kaveggia, E. G. Studies of malformation syndromes of man 33: the FG syndrome. An X-linked recessive syndrome of multiple congenital anomalies and mental retardation. Z. Kinderheilkd 117, 1–18 (1974).
Lujan, J. E., Carlin, M. E. & Lubs, H. A. A form of X-linked mental retardation with marfanoid habitus. Am. J. Med. Genet. 17, 311–322 (1984).
Schwartz, C. E. et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J. Med. Genet. 44, 472–477 (2007).
Maat-Kievit, A., Brunner, H. G. & Maaswinkel-Mooij, P. Two additional cases of the Ohdo blepharophimosis syndrome. Am. J. Med. Genet. 47, 901–906 (1993).
Vulto-van Silfhout, A. T. et al. Mutations in MED12 cause X-linked Ohdo syndrome. Am. J. Hum. Genet. 92, 401–406 (2013).
Bouazzi, H., Lesca, G., Trujillo, C., Alwasiyah, M. K. & Munnich, A. Nonsyndromic X-linked intellectual deficiency in three brothers with a novel MED12 missense mutation [c.5922G>T (p.Glu1974His)]. Clin. Case. Rep. 3, 604–609 (2015).
Lesca, G. et al. Clinical and neurocognitive characterization of a family with a novel MED12 gene frameshift mutation. Am. J. Med. Genet. A 161A, 3063–3071 (2013).
Rump, P. et al. A novel mutation in MED12 causes FG syndrome (Opitz–Kaveggia syndrome). Clin. Genet. 79, 183–188 (2011).
Charzewska, A. et al. The power of the Mediator complex—expanding the genetic architecture and phenotypic spectrum of MED12-related disorders. Clin. Genet. 94, 450–456 (2018).
Donnio, L. M. et al. MED12-related XLID disorders are dose-dependent of immediate early genes (IEGs) expression. Hum. Mol. Genet. 26, 2062–2075 (2017).
Hu, H. et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 21, 133–148 (2016).
Langley, K. G. et al. Beyond Ohdo syndrome: a familial missense mutation broadens the MED12 spectrum. Am. J. Med. Genet. A 167A, 3180–3185 (2015).
Narayanan, D. L. & Phadke, S. R. A novel variant in MED12 gene: further delineation of phenotype. Am. J. Med. Genet. A. 173, 2257–2260 (2017).
Niranjan, T. S. et al. Affected kindred analysis of human X chromosome exomes to identify novel X-linked intellectual disability genes. PLoS ONE 10, e0116454 (2015).
Patil, S. J. et al. Clinical variability in familial X-linked Ohdo syndrome-Maat-Kievit-Brunner type with MED12 mutation. J. Pediatr. Genet. 6, 198–204 (2017).
Prescott, T. E. et al. Two male sibs with severe micrognathia and a missense variant in MED12. Eur. J. Med. Genet. 59, 367–372 (2016).
Prontera, P. et al. A novel MED12 mutation: evidence for a fourth phenotype. Am. J. Med. Genet. A. 170, 2377–2382 (2016).
Srivastava, S. et al. Dysregulations of sonic hedgehog signaling in MED12-related X-linked intellectual disability disorders. Mol. Genet. Genomic. Med. 7, e00569 (2019).
Tzschach, A. et al. Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 23, 1513–1518 (2015).
Yamamoto, T. & Shimojima, K. A novel MED12 mutation associated with nonspecific X-linked intellectual disability. Hum. Genome. Var. 2, 15018 (2015).
Murakami, H., Enomoto, Y., Tsurusaki, Y., Sugio, Y. & Kurosawa, K. A female patient with X-linked Ohdo syndrome of the Maat-Kievit-Brunner phenotype caused by a novel variant of MED12. Congenit Anom (Kyoto). 60, 91–93 (2020).
Fieremans, N. et al. Identification of intellectual disability genes in female patients with a skewed X-inactivation pattern. Hum. Mutat. 37, 804–811 (2016).
Wang, C. et al. MED12-related disease in a Chinese girl: clinical characteristics and underlying mechanism. Front Genet 11, 129 (2020).
Firth, H. V. et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 84, 524–533 (2009).
Sobreira, N., Schiettecatte, F., Valle, D. & Hamosh, A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–930 (2015).
Allen, R. C., Zoghbi, H. Y., Moseley, A. B., Rosenblatt, H. M. & Belmont, J. W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 51, 1229–1239 (1992).
Brogna, S. & Wen, J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 16, 107–113 (2009).
Zweier, C. et al. A new face of Borjeson-Forssman-Lehmann syndrome? De novo mutations in PHF6 in seven females with a distinct phenotype. J. Med. Genet. 50, 838–847 (2013).
Lyons, M. J. et al. Clinical experience in the evaluation of 30 patients with a prior diagnosis of FG syndrome. J. Med. Genet. 46, 9–13 (2009).
Risheg, H. et al. A recurrent mutation in MED12 leading to R961W causes Opitz–Kaveggia syndrome. Nat. Genet. 39, 451–453 (2007).
Verloes, A. et al. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am. J. Med. Genet. A 140, 1285–1296 (2006).
Isidor, B. et al. Blepharophimosis, short humeri, developmental delay and hirschsprung disease: expanding the phenotypic spectrum of MED12 mutations. Am. J. Med. Genet. A 164A, 1821–1825 (2014).
Acknowledgements
We are grateful to the patients and their families for their participation. This work was supported by the European Union’s Seventh Framework Program (Gencodys; grant 241995 to H.v.B). D.L.P. is recipient of a CAPES Fellowship (99999.013311/2013–01). C.Z. was supported by grants from the German Research Foundation (DFG) (ZW184/3–1, ZW184/6–1 and 270949263/GRK2162) and by the IZKF Erlangen (E31). B.B.A.d.V was supported from the Dutch Organization for Health Research and Development (ZON-MW grants 917–86–319 and 912–12–109). C.T.R.M.S. and A.P.A.S. are supported by the European Reference Network (ERN) ITHACA (project ID no 739543). The 100,000 Genomes Project is funded by the National Institute of Health Research and NHS England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research structure. This study makes use of data generated by the Genomics England Research Consortium, Deciphering Developmental Disorders (DDD) study, and the DECIPHER community. A full list of centers that contributed to the generation of the data is available from https://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by Wellcome Trust.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.d.B., A.R. C.Z., H.v.B. Data curation: A.d.B, C.Z. D.L.P. Formal analysis: D.L.P. Funding acquisition: A.P.A.S., B.B.A.d.V, C.T.R.M.S., C.Z., D.L.P., H.v.B. Investigation: A.G., A.P.A.S., A.T.V., B.B.A.d.V., C.D., C.T.R.M.S., C.Z., D.L., D.L.P., E.J.B., E.K.S.M.L., E.M.H.F.B., E.Z., H.H., G.G., J.B.G.M.V., J.S.K., K.C., K.H-K., L.G., N.R., R.W., S.A.L., S. Banka, S. Berland, S.C., S.C.H., W.R. Methodology: R.P. Supervision: A.d.B., A.R., C.Z. H.v.B. Visualization: A.d.B., C.Z., D.L.P. Writing: A.d.B., C.Z., D.L.P. Writing (review and editing): A.d.B., C.Z., D.L.P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declaration
Individuals were identified in different centers worldwide in diagnostic or research settings approved by the respective institutional review boards in each case. The study as a whole was approved by the review board of the Friedrich-Alexander-University Erlangen-Nürnberg. Written informed consent was obtained for all individuals involved. For publishing the patients’ photos, specific written informed consent was obtained as well. This study adhered to the World Medical Association Declaration of Helsinki (2013).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Polla, D.L., Bhoj, E.J., Verheij, J.B.G.M. et al. De novo variants in MED12 cause X-linked syndromic neurodevelopmental disorders in 18 females. Genet Med 23, 645–652 (2021). https://doi.org/10.1038/s41436-020-01040-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01040-6
This article is cited by
-
Mutation spectrum of Kallmann syndrome: identification of five novel mutations across ANOS1 and FGFR1
Reproductive Biology and Endocrinology (2023)
