
Abstract
Purpose
Metachromatic leukodystrophy (MLD) is a lysosomal storage disorder caused by the deficiency of arylsulfatase A (ARSA), which results in the accumulation of sulfatides. Newborn screening for MLD may be considered in the future as innovative treatments are advancing. We carried out a research study to assess the feasibility of screening MLD using dried blood spots (DBS) from de-identified newborns.
Methods
To minimize the false-positive rate, a two-tier screening algorithm was designed. The primary test was to quantify C16:0-sulfatide in DBS by ultraperformance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS). The screening cutoff was established based on the results from 15 MLD newborns to achieve 100% sensitivity. The secondary test was to measure the ARSA activity in DBS from newborns with abnormal C16:0-sulfatide levels. Only newborns that displayed both abnormal C16:0-sulfatide abundance and ARSA activity were considered screen positives.
Results
A total of 27,335 newborns were screened using this two-tier algorithm, and 2 high-risk cases were identified. ARSA gene sequencing identified these two high-risk subjects to be a MLD-affected patient and a heterozygote.
Conclusion
Our study demonstrates that newborn screening for MLD is highly feasible in a real-world scenario with near 100% assay specificity.
Similar content being viewed by others

INTRODUCTION
Metachromatic leukodystrophy (MLD, OMIM 250100) is a lysosomal storage disorder where the desulfation of 3-sulfogalactosylceramides (sulfatides) is impaired. The desulfation of sulfatides requires the combined action of arylsulfatase A (ARSA) and its activator protein, saposin B. Therefore, MLD is caused by the deficiency of ARSA, or more rarely, saposin B.1,2 MLD is characterized clinically by symptoms related to progressive demyelination and is classified according to age of onset as late-infantile (<30 months), juvenile (2.5 to 16 years) and adult (>16 years).2 Hematopoietic stem cell transplantation can benefit children with presymptomatic late-infantile MLD or minimally symptomatic juvenile MLD.3 Innovative treatments, including ex vivo gene therapy and enzyme replacement therapy, for MLD are being evaluated in ongoing clinical trials.4 Given these treatments, newborn screening (NBS) for MLD is being considered.3
Biochemical assays using dried blood spots (DBS) have been developed to support NBS of MLD using various strategies, including the quantification of (1) sulfatides, (2) ARSA protein abundance (immunoassays), and most recently (3) ARSA activity.5,6,7 Screening for MLD based on the ARSA activity was not previously considered because a DBS-based assay was not available until recently; the thermal instability of ARSA together with the high frequency of pseudodeficiency variants in the normal population poses additional challenges.6 Ridsdale et al. conducted a screening study of nearly 100,000 de-identified newborns using an immunoassay to quantify the abundance of ARSA in DBS.8 A total of 73 newborns had abnormally low ARSA protein abundance in DBS, none of whom were considered at risk after ARSA gene analysis and sulfatide measurement.8 However, the sensitivity of this immunoassay is questionable as it could give rise to false-negative errors in the case of properly folded, but enzymatically deficient proteins.9 Moreover, the specificity of this immunoassay is also compromised because one of the most common ARSA pseudodeficiency variants, c.*96A>G, causes the reduction of ARSA messenger RNA (mRNA) and hence ARSA protein by about 90% due to the polyadenylation defect.10,11 On the contrary, it was reported that MLD newborns (newborns who developed MLD later in life) had elevated sulfatide level in blood when compared with individuals with two ARSA pseudodeficiency variants in trans and with normal newborns.5 This suggests that sulfatide analysis is a better screening test than an immunoassay.
Our laboratory initiated a research study for NBS of MLD by measuring sulfatide abundance in DBS from de-identified newborns. When sulfatides were elevated above the cutoff, we used another punch from the same DBS to measure ARSA enzymatic activity. In principle, this two-tier screening strategy could also identify patients with multiple sulfatase deficiency (MSD), where the defect is in the formylglycine generating enzyme (FGE).12 FGE, encoded by SUMF1, is required to activate all cellular sulfatases, including ARSA, therefore MSD patients also display high sulfatide and low ARSA activity in blood.12
MATERIALS AND METHODS
DBS samples
This study using DBS from de-identified newborns was approved by the Washington State Institutional Review Board. The DBS were shared by the Washington State Department of Health after being stored for 30–60 days at room temperature. A total of 15 archived DBS from MLD newborns (10 late-infantile and 5 juvenile onset) were acquired through the MLD Foundation, the University of Pittsburgh, and the Meyer Children’s Hospital (Florence, Italy). One of the 15 MLD newborns was diagnosed with saposin B deficiency (MLD newborn 6, Table S1). Results from the random newborns and the 15 MLD newborns are summarized in Table S1.
Methods
The detailed protocol for the assay is provided in the Supplementary Information. In short, sulfatides from a 3-mm DBS punch were extracted with methanol containing the internal standard (d5-C16:0-sulfatide). Methanol containing C16:0-sulfatide (external calibrators) was also included in each plate in triplicate and processed at the same time as the DBS. After a 4-hour extraction, the sample was centrifuged, and the supernatant was analyzed by ultraperformance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS), where the analyte and its internal standard were detected by multiple reaction monitoring in electrospray ionization (ESI) negative mode. Newborns with normalized C16:0-sulfatide level above 0.64 were considered at risk of MLD and were subjected to a second-tier test.
A second 3-mm punch from newborns with sulfatide level above the screening cutoff, along with punches from 6 to 8 newborns with normal sulfatide level and similar storage condition (matching newborns), were submitted to the ARSA enzymatic assay using the reported protocol.7 For newborns with sulfatide level above the cutoff (0.64 normalized C16:0-sulfatide) and ARSA activity below the cutoff (20% to the mean activity of the matching newborns), activities of iduronate-2-sulfatase (I2S), N-acetylgalactosamine-6-sulfatase (GALNS), and N-acetylgalactosamine-4-sulfatase (ARSB) were measured as previously described.13 Newborns with abnormal sulfatide levels and ARSA activity but normal I2S, GALNS, and ARSB activities were considered as MLD screen positives. Newborns with abnormal sulfatide levels and ARSA, I2S, GALNS, and ARSB activities were considered as MSD screen positives. Genetic sequencing of the ARSA gene or the SUMF1 gene was performed on a third 3-mm DBS punch accordingly.
Methods used for the ARSA gene sequencing and the subsequent data analysis are provided in the Supplementary Information.
Ethics statement
This study using DBS from de-identified newborns was approved by the Washington State Institutional Review Board.
RESULTS
UPLC-MS/MS analysis of sulfatide in DBS
In our previous study, it was observed that four sulfatide species were elevated in DBS from MLD patients compared with healthy controls.5 C16:0- and C16:0-OH-sulfatide (named according to the fatty acyl group) account for more than 85% of the sulfatides in blood, whereas C16:1-OH- and C18:0-sulfatide account for the remainder.5 Therefore, we initiated a research study of NBS of MLD by monitoring the total amount of these four sulfatide species in DBS using high throughput UPLC-MS/MS. Studies on assay performance were carried out in our previous study and therefore were not repeated herein.5 Fig. 1a shows a typical UPLC-MS/MS chromatogram of C16:1-OH, C16:0-OH, C16:0, and C18:0-sulfatide in DBS from a random newborn. The sample injection-to-injection time was 2.5 minutes, allowing more than 500 samples to be analyzed per day per instrument.

a UPLC-MS/MS chromatogram of four sulfatide species in DBS from a random newborn. The x-axis is time (min) and the y-axis is the MS/MS intensity. Peak 1 at 1.22 min is C16:1-OH-sulfatide, peak 2 at 1.31 min is C16:0-OH-sulfatide, peak 3 at 1.35 min is C16:0-sulfatide and peak 4 at 1.46 min is C18:0-sulfatide. b C16:0-sulfatide abundance (μM) in DBS from 15 MLD newborns (median: 0.32 μM, range: 0.18–0.47 μM) and 2000 random newborns (median: 0.094 μM, range: 0.020–0.23 μM). The solid line is the median of the MLD group. The dash line is the screening cutoff at 0.17 μM. The crosses indicate MLD newborns without normalized C16:0-sulfatide data available (see text). The circles indicate MLD newborns with normalized C16:0-sulfatide data available and the data are presented in (c). c Normalized C16:0-sulfatide level in DBS from 6 MLD newborns (median: 1.24, range: 0.68–0.48) and 2000 random newborns (median: 0.34, range: 0.11–0.86). The dash line is the screening cutoff at 0.64 after normalization.
Sulfatide analysis in DBS from MLD newborns
DBS from a total of 15 MLD newborns were acquired from NBS laboratory repositories. Individual molecular sulfatide species levels in DBS and age of onset information are given in Table S1. Two patients with juvenile MLD displayed substantially lower sulfatide level compared with the rest of the cohort (MLD newborn 12 and 13, Table S1). The sulfatide assay was repeated on 2 additional punches from the same DBS from these patients with the same results. An ARSA protein abundance assay and an ARSA enzymatic activity assay were also performed, with essentially no ARSA protein and ARSA activity detected (data not shown). These results suggest that the correct archived DBS were pulled out of storage. These archived DBS were stored at −20 °C. Sulfatides should be relatively stable under this storage condition for at least 1 year based on our experience with adult DBS. Elevation in blood sulfatides was observed in a MLD newborn with late-infantile onset caused by saposin B deficiency, which was consistent with reports (MLD newborn 6, Table S1).2,14
Analysis of total sulfatide in DBS
Figure S1 shows the total sulfatide abundance in 15 MLD newborns (median: 0.79 µM, range: 0.50–1.23 µM) and 2,000 random newborns (median: 0.25 μM, range: 0.085–0.56 µM). However, problems were encountered in total sulfatide measurement after a new UPLC column was installed. The retention times for the four sulfatides decreased initially by about 0.4 minute but then shifted gradually to higher times after repeated sample injections. The retention times stabilized after about 1,000 injections. Similar retention time shifts were observed when using two additional UPLC columns with different solid phase chemistry (data not shown). As a result, a discontinuous change in the apparent total sulfatide level in DBS was observed after a new column was installed (Fig. S2).
These shifts in retention time were concerning because a universal internal standard (d5-C16:0-sulfatide) was used for the four endogenous sulfatide species eluting at different retention times. A shift in retention time will result in a change in ionization suppression. This change can only be accounted for by the use of a chemically identical but isotopically substituted internal standard such that the analyte and its internal standard coelute from the column and are suppressed to the same extent. Therefore, a second internal standard, d3-C16:0-OH-sulfatide, was added to the assay, since C16:0-OH-sulfatide and C16:0-sulfatide accounted for the majority of the sulfatides in DBS (Fig. 1a).5 However, endogenous isobaric interferences from DBS was observed in the detection channel for d3-C16:0-OH-sulfatide. Chromatographic separation of these isobars from the additional internal standard, d3-C16:0-OH-sulfatide, required a lengthened analysis that would lower the throughput of the method. Therefore, we discontinued the measurement of total sulfatides in DBS and started to monitor the single sulfatide species C16:0-sulfatide using d5-C16:0-sulfatide as the internal standard. This was justified by the fact that C16:0-sulfatide is a major sulfatide in DBS and was elevated in DBS from the 15 MLD newborns to a similar extent as total sulfatides (Table S1).
Analysis of C16:0-sulfatide in DBS
Figure1b displays the C16:0-sulfatide concentration in DBS from 15 MLD newborns (median: 0.32 µM, range: 0.18–0.47 µM) and 2,000 random newborns (median: 0.094 µM, range: 0.020–0.23 µM). A distribution overlap between the MLD newborns and the random newborns was observed (Fig. 1b). C16:0-sulfatide of 0.17 µM was used as the screening cutoff to achieve 100% assay sensitivity.
To further harmonize the results, for every 96-well plate of newborn DBS, we carried out 3 separate UPLC-MS/MS analyses of an authentic C16:0-sulfatide standard processed the same as the newborn DBS. The C16:0-sulfatide/internal standard ion ratio measured for each newborn was divided by the averaged ratio measured for the 3 injections of the C16:0-sulfatide standard (normalization method). Shown in Fig. S3 is the C16:0-sulfatide level in groups of 500 newborns collected over 5 months with or without normalization. Results with normalization were more consistent, suggesting that the variations were mostly introduced from internal standard batch-to-batch differences as well as fluctuations of the mass spectrometer, which could be accounted for by the normalization to the external calibrator.
Our final approach for the screening of MLD was to quantify C16:0-sulfatide in DBS using d5-C16:0-sulfatide as the internal standard and then to normalize to the external calibrator. Figure 1c displays the normalized C16:0-sulfatide level in DBS from 6 MLD newborns (median: 1.24, range: 0.68–1.48) and 2,000 random newborns (median: 0.34, range: 0.11–0.86). We did not have data on all the 15 MLD newborns due to the limited patient samples. However, we did have normalized C16:0-sulfatide results from the 2 MLD newborns displaying the lowest sulfatide abundance (MLD newborn 12 and 13, Table S1), which allowed us to define the cutoff to achieve 100% sensitivity. The screening cutoff was set at 0.64 after the normalization, which corresponds to 0.17 µM C16:0-sulfatide in blood (Fig. 1c).
A total of 27,335 random newborns were screened using this strategy, of which 195 (0.71%) had normalized C16:0-sulfatide level above the cutoff (Table 1). The distributions of the C16:0-sulfatide abundance in the 27,335 newborns with or without the normalization are shown in Fig. S4. Results of the 195 newborns with abnormal C16:0-sulfatide level are summarized in Table S2.
Implementation of ARSA DBS activity assay as a second-tier test
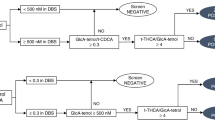
Given that the C16:0-sulfatide analysis resulted in a 0.71% screen-positive rate, an ARSA DBS enzymatic activity assay was implemented as the second-tier test using an additional 3-mm punch from the same DBS that was used for sulfatide analysis (algorithm shown in Fig. 2a). Since ARSA is known to be unstable in DBS at room temperature, ARSA activity was also measured in “matching newborns” with normal sulfatide levels and similar storage conditions to define a reference range.6,7 If ARSA was low, the activities of three additional sulfatases (I2S, GALNS, and ARSB) were measured to further distinguish MLD screen positives from MSD screen positives (Fig. 2a).

a The C16:0-sulfatide DBS assay is the primary screening test and the ARSA DBS activity assay is the secondary test; b the ARSA DBS activity assay is the primary screening test and the C16:0-sulfatide DBS assay is the secondary test.
Among those 195 newborns with C16:0-sulfatide abundance above 0.64 after normalization, 122 were submitted for ARSA activity assay. The rest were not tested for ARSA activity as the DBS were too old (more than 3 months at room temperature). All but two newborns with abnormal C16:0-sulfatide level had ARSA activity above 20% relative to their matching newborns, showing that they were all false positives identified by the sulfatide assay (Table 1, Table S2). Subject 24 (Table S2) displayed C16:0-sulfatide at 0.86 and 0% ARSA activity, and subject 128 (Table S2) displayed C16:0-sulfatide at 0.72 and 8% ARSA. Activities of I2S, GALNS, and ARSB were only measured in subject 128 (Table S2), with the activities all above 20% of the mean activities, ruling out MSD. Even though there were no I2S, GALNS and ARSB activity data on subject 24 (Table S2), reserved DBS punches from these two newborns were both submitted for ARSA gene sequencing, since MSD is much rarer than MLD.2,12
ARSA genomic variant analysis
ARSA exome sequencing was performed on five DBS samples for variant discovery. The samples consisted of two MLD screen positives (subjects 24 and 128, Table S2) and three MLD screen negatives (elevated sulfatides but high ARSA enzymatic activity, subjects 9, 11, and 23, Table S2). Sequencing results are summarized in Table 2. Three variants, including one exonic (c.1178C>G) and two intronic (c.1210+20C>G and c.1108-32C>T), were found across all five samples (Table 2). c.1178C>G is a known pseudodeficiency variant.15,16 c.1210+20C>G is classified as benign in ClinVar. c.1108-32C>T is not reported in ClinVar; it has higher than expected population allele frequencies and was therefore not considered to be pathogenic (0.7979, gnomAD). Additionally, one screen-positive and two screen negative samples had the common pseudodeficiency variant c.1055A>G (p.Asn352Ser) (Table 2).1 Pathogenic variants were only discovered in the two screen-positive samples (Table 2). The status of “screen negative” of subjects 9, 11, and 23 (Table S2) was confirmed by ARSA sequencing (Table 2).
Subject 24 (Tables 2 and S2) was heterozygous for the pathogenic variants c.1283C>T and c.1292A>C.1,17 Although ClinVar does not have an interpretation for c.1292A>C (previously known as c. 1286A>C), this variant has been listed as pathogenic in multiple reports.1,17 This subject was also heterozygous for the common pseudodeficiency variant, c.1178C>G.15,16 Therefore, subject 24 was interpreted as an MLD-affected patient.
Subject 128 (Tables 2 and S2) was heterozygous for c.1174C>T (previously known as c. 1168C>T). ClinVar has conflicting interpretations of pathogenicity, but it is deemed as pathogenic according to multiple literature reports.1,18,19,20,21 This subject is also heterozygous and homozygous for the pseudodeficiency variants c.1178C>G and c.1055A>G, respectively.1 Taken together, subject 128 was interpreted as a heterozygote.
ARSA DBS activity assay as the first-tier screening test for MLD
Since the sulfatide assay resulted in a high false-positive rate (0.71%), we explored if the ARSA DBS activity assay could suffice as the primary screening test. Shown in Fig. 2b is the proposed screening algorithm, where the C16:0-sulfatide DBS assay was implemented as the secondary test. The selection of the 20% daily mean activity cutoff was based on our previously reported data.7
ARSA activity in DBS from 2,287 de-identified newborns was measured using a reported protocol.7 Three of the 2287 newborns (0.13%) had ARSA activity below the cutoff, all of which were considered screen negative based on the C16:0-sulfatide results (Tables 1, S3). Results from the 2,287 newborns and the three subjects with low ARSA activity are summarized in Table S3.
During summer in warm states like Florida and Texas, the transport conditions of DBS can be relatively harsh. Thus, the stability of ARSA under ambient conditions was assessed by measuring its activity in DBS stored at room temperature and at 37 °C with different relative humidity settings for a period of up to 7 days. As shown in Fig. S5, less than 50% of the ARSA activity was lost in the first three days under all conditions. The activity continued to decrease, with 25% remaining after the DBS were stored at 37 °C with a 33% relative humidity for 7 days (Fig. S5). Intriguingly, it appeared that a higher relative humidity helped to preserve ARSA in DBS, as over 50% ARSA activity remained after the DBS were stored at 37 °C with a 84% relative humidity for 7 days (Fig. S5).
DISCUSSION
This study demonstrates that even a slight difference in the biomarker level in DBS between disease-affected patients and healthy controls is sufficient to develop a successful NBS method. MLD newborns can be distinguished from normal newborns based on C16:0-sulfatide abundance in DBS with statistical significance. To maintain assay robustness, it requires the use of an isotope-labeled but chemically identical internal standard (d5-C16:0-sulfatide) and external calibration on a routine basis to allow normalization. Having an internal standard that coelutes with the analyte is critical as it negates variations brought by chromatographic changes (matrix effects).
DBS-based second-tier tests are often implemented as part of routine screening, which reduces the false-positive rates of primary tests.22,23,24,25 This greatly reduces family anxiety as well as the burden to the medical system. The ARSA enzymatic activity assay is done with the same UPLC-MS/MS equipment as the sulfatide analysis and thus does not add substantially to the cost of MLD NBS. However, MLD patients due to saposin B deficiency would be missed by our algorithm as they would display abnormal sulfatide level but near-normal ARSA activity.1 If patients with saposin B deficiency are of interest, an immunoassay for saposin B can be implemented as a secondary test to follow up newborns with abnormal sulfatide abundance. Such an assay has been reported using plasma as the sample specimen.26 Nevertheless, although it was reported that bone marrow transplantation could benefit saposin B patients, this condition is of lower priority to NBS as it is much rarer than MLD caused by ARSA deficiency.1,27 Futhermore, gene therapy for MLD is based on ARSA expression and would not be useful to treat saposin B–dependent MLD.
We did not have good coverage for the other common pseudodeficiency variant, c.*96A>G, which was reported to reduce ARSA activity to 10% of control in vivo.10,11 Even though c.*96A>G and c.1055A>G (p.Asn352Ser) usually occur in cis, it was also found that c.1055A>G could appear independently, especially in Africans and East Asians.1,28 Therefore, the appearance of c.1055A>G in subjects 128, 11, and 23 (Table 2) strongly suggests the presence of c.*96A>G but is not confirmative.
Although it is well known that subjects with an ARSA pseudodeficiency variant in trans with a pathogenic ARSA variant do not result in MLD, Hohenschutz et al. reported a patient with a compound heterozygous genotype of ARSA pathogenic and pseudodeficiency variantions, who displayed intermediate ARSA activity between adult MLD and subjects with pseudodeficiency, slightly elevated urine sulfatide, and more importantly, neuropsychiatric symptoms.2,29 This patient developed the first symptom at the age of 36 and did not deteriorate until 16 years later.29 Hence, it is possible that subject 128 (Table 2, Table S2), an apparent MLD heterozygote, will display some mild symptoms later in life.
Two screen positives were identified from 27,335 newborns, among which one is a MLD-affected patient while the other is a likely heterozygote, thus highlighting adequate specificity of the two-tier screening algorithm.30,31,32 The false-negative rate of the study is hard to assess. Yet considering the prevalence of MLD (1:40,000 to 1:160,000) and the fact that 1 patient was identified out of 27,335 newborns, it is probable that no patients were missed.2
False negatives can only be addressed by surveillance of patient reports, and this is possible in the future since the approximate birthdates of the newborns tested in our study are known. The screening cutoff for the C16:0-sulfatide assay was based on the results from the 15 MLD newborns to achieve 100% sensitivity. Obtaining more DBS from MLD newborns can help to better assess the cutoff selection and to gauge whether the two newborn MLD patients with lower sulfatides than the rest are anomalous or representative. If we omit these two newborn samples, the false-positive rate using first-tier C16:0-sulfatide would decrease to 0.19%, therefore still necessitating a second-tier test. Hopefully, with increasing awareness of MLD and future prospective screening studies, more specimens from MLD newborns will become available. The cutoff (20% daily mean activity) for the ARSA DBS activity assay was based on results from a previous report, and was more conservative compared with other enzymatic assays for lysosomal enzymes.7,31,33
To date, lysosomal storage diseases that are due to malfunctioning enzymes are typically screened using functional assays; however, ARSA DBS activity assay was not available until recently. This screening strategy was further discredited due to two major concerns: (1) the potential false-positive problems caused by high prevalence of pseudodeficiency variants and (2) the thermal instability of ARSA in DBS.6 Nonetheless, given the high false-positive rate (0.71%) of the sulfatide assay, ARSA activity assay may suffice as the primary test while using the C16:0-sulfatide assay as a secondary test (Fig. 2b). This is supported by the lower false-positive rate (0.13%) from the small-scale study on 2,287 newborns (Table 1). As shown in Fig. S4, 50% of the ARSA activity remained when the DBS were stored under extreme conditions for three days. Since DBS should be delivered to the screening laboratories within 3 days of sample collection, the ARSA thermal instability may not be a major issue; however, it remains to be seen how problematic this will be in warmer parts of the world. Currently the ARSA assay is more complex than the sulfatide assay; nevertheless, with automation the ARSA activity and sulfatide assays can be carried out by a single laboratory worker and multiplexed with other assays.
In conclusion, the large-scale study presented here demonstrates that NBS for MLD is feasible with an exceptionally low false-positive rate (0.0037%) if a two-tier screening strategy is adopted. We propose that newborns be screened based on the abundance of blood sulfatide and ARSA enzymatic activity be measured in those with abnormal sulfatide results. This two-tier screening approach is crucial for the balance between assay sensitivity and specificity. A total of 27,335 newborns were screened with this approach, and only 2 of which were considered screen positives. ARSA gene sequencing identified one as an MLD patient and the other as heterozygous, demonstrating that this two-tier strategy is highly specific. Alternatively, it may also be possible to screen for MLD using the ARSA activity assay as the primary test, and the sulfatide assay as the secondary test. Implementation of these two-tier screening strategies will be useful in identifying patients who will then benefit from upcoming novel therapies.
References
Cesani, M. et al. Mutation update of ARSA and PSAP genes causing metachromatic leukodystrophy. Hum. Mutat. 37, 16–27 (2016).
Gieselmann, V. & Ingeborg, K.-M. Metachromatic Leukodystrophy. In The Online Metabolic and Molecular Bases of Inherited Disease, (eds Valle, D. L., Antonarakis, S. Ballabio, A., Beaudet, A. L. & Mitchell, G. A.) (McGraw-Hill Education: New York, NY, 2019).
Martin, H. R. et al. Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy. Biol. Blood Marrow Transplant. 19, 616–624 (2013).
Poletti, V. & Biffi, A. Gene-based approaches to inherited neurometabolic diseases. Hum. Gene Ther. 30, 1222–1235 (2019).
Spacil, Z. et al. Sulfatide analysis by mass spectrometry for screening of metachromatic leukodystrophy in dried blood and urine samples. Clin. Chem. 62, 279–286 (2016).
Tan, M. A., Dean, C. J., Hopwood, J. J. & Meikle, P. J. Diagnosis of metachromatic leukodystrophy by immune quantification of arylsulphatase A protein and activity in dried blood spots. Clin. Chem. 54, 1925–1927 (2008).
Hong, X. et al. Leukocyte and dried blood spot arylsulfatase A assay by tandem mass spectrometry. Anal. Chem. 92, 6341–6348 (2020).
Ridsdale, R. et al. Newborn screening (NBS) for metachromatic leukodystrophy (MLD): results from a study of 100,000 deidentified NBS samples. Abstr. Mol. Genet. Metab. 120, S17–S145 (2016).
Umapathysivam, K. et al. Determination of acid α-glucosidase protein: evaluation as a screening marker for Pompe disease and other lysosomal storage disorders. Clin. Chem. 46, 1318–1325 (2000).
Harvey, J. S., Carey, W. F. & Morris, C. P. Importance of the glycosylation and polyadenylation variants in metachromatic leukodystrophy pseudodeficiency phenotype. Hum. Mol. Genet. 7, 1215–1219 (1998).
Gieselmann, V., Polten, A., Kreysing, J. & Von Figura, K. Arylsulfatase A pseudodeficiency: loss of a polyadenylylation signal and N-glycosylation site. Proc. Natl. Acad. Sci. A 86, 9436–9440 (1989).
Hopwood, J. J. & Ballabio, A. Multiple Sulfatase Deficiency and the Nature of the Sulfatase Family. In The Online Metabolic and Molecular Bases of Inherited Disease, (eds Valle, D. L., Antonarakis, S., Ballabio, A., Beaudet, A. L. & Mitchell, G. A.) (McGraw-Hill Education, New York, NY, 2019).
Liu, Y. et al. Multiplex tandem mass spectrometry enzymatic activity assay for newborn screening of the mucopolysaccharidoses and type 2 neuronal ceroid lipofuscinosis. Clin. Chem. 63, 1118–1126 (2017).
Wenger, D. A. et al. Clinical, pathological, and biochemical studies on an infantile case of sulfatide/GM1 activator protein deficiency. Am. J. Med. Genet. 33, 255–265 (1989).
Gomez-Ospina N. Arylsulfatase A Deficiency. 2006 May 30 [Updated 2020 Apr 30]. In GeneReviews® (eds Adam M. P. et al.) [Internet]. (University of Washington, Seattle, 1993-2020).
Hettiarachchi, D. & Dissanayake, V. H. W. Three novel variants in the arylsulfatase A (ARSA) gene in patients with metachromatic leukodystrophy (MLD). BMC Res. Notes 12, 726 (2019).
Eng, B. et al. Identification of nine novel arylsulfatase a (ARSA) gene mutations in patients with metachromatic leukodystrophy (MLD). Hum. Mutat. 22, 418–419 (2003).
Shukla, P. et al. Molecular and structural analysis of metachromatic leukodystrophy patients in Indian population. J. Neurol. Sci. 301, 38–45 (2011).
Wolf, N. I. et al. Metachromatic leukodystrophy and transplantation: remyelination, no cross-correction. Ann. Clin. Transl. Neurol. 7, 169–180 (2020).
Narayanan, D. L. et al. Spectrum of ARSA variations in Asian Indian patients with arylsulfatase A deficient metachromatic leukodystrophy. J. Hum. Genet. 64, 323–331 (2019).
Barth, M. L., Fensom, A. & Harris, A. Identification of seven novel mutations associated with metachromatic leukodystrophy. Hum. Mutat. 6, 170–176 (1995).
Chace, D. H. & Hannon, W. H. Impact of second-tier testing on the effectiveness of newborn screening. Clin. Chem. 56, 1653–1655 (2010).
Langan, T. J. et al. Development of a newborn screening tool based on bivariate normal limits: using psychosine and galactocerebrosidase determination on dried blood spots to predict Krabbe disease. Genet. Med. 21, 1644–1651 (2018).
Tortorelli, S. et al. Moonlighting newborn screening markers: the incidental discovery of a second-tier test for Pompe disease. Genet. Med. 20, 840–846 (2018).
Turgeon, C. T. et al. Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin. Chem. 56, 1686–1695 (2010).
Chang, M. H. et al. Saposins A, B, C, and D in plasma of patients with lysosomal storage disorders. Clin. Chem. 46, 167–174 (2000).
Krivit, W. Allogeneic stem cell transplantation for the treatment of lysosomal and peroxisomal metabolic diseases. Springer Semin. Immunopathol. 26, 119–132 (2004).
Ott, R., Waye, J. S., Chang, P. L. & Chang, P. Evolutionary origins of two tightly linked mutations in arylsulfatase-A pseudodeficiency. Hum. Genet. 101, 135–140 (1997).
Hohenschutz, C. et al. Probable metachromatic leukodystrophy/pseudodeficiency compound heterozygote at the arylsulfatase A locus with neurological and psychiatric symptomatology. Am. J. Med. Genet. 31, 169–175 (1988).
Hopkins, P. V. et al. Incidence of 4 lysosomal storage disorders from 4 years of newborn screening. JAMA Pediatr. 172, 696–697 (2018).
Burton, B. K. et al. Newborn screening for lysosomal storage disorders in Illinois: the initial 15-month experience. J. Pediatr. 190, 130–135 (2017).
Lee, S. et al. Evaluation of X-linked adrenoleukodystrophy newborn screening in North Carolina. JAMA Netw. Open 3, e1920356 (2020).
Scott, C. R. et al. Newborn screening for mucopolysaccharidoses: results of a pilot study with 100 000 dried blood spots. J. Pediatr. 216, 204–207 (2019).
Acknowledgements
We are grateful for Giancarlo la Marca for providing part of the MLD newborn DBS used in the study. Funding is provided by Takeda and National Institutes of Health (R01 DK067859).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
M.H.G. is a consultant for PerkinElmer Corp. PerkinElmer was not involved in any aspects of the study described in this paper. The other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Hong, X., Daiker, J., Sadilek, M. et al. Toward newborn screening of metachromatic leukodystrophy: results from analysis of over 27,000 newborn dried blood spots. Genet Med 23, 555–561 (2021). https://doi.org/10.1038/s41436-020-01017-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01017-5
This article is cited by
-
A systematic review on the birth prevalence of metachromatic leukodystrophy
Orphanet Journal of Rare Diseases (2024)
-
Inventory of current practices regarding hematopoietic stem cell transplantation in metachromatic leukodystrophy in Europe and neighboring countries
Orphanet Journal of Rare Diseases (2024)
-
Predicting disease severity in metachromatic leukodystrophy using protein activity and a patient phenotype matrix
Genome Biology (2023)
-
Entwicklung der Analytik im Neugeborenen-Screening – Von der Guthrie-Karte zur Genetik
Bundesgesundheitsblatt - Gesundheitsforschung - Gesundheitsschutz (2023)
-
Methods and feasibility study for exome sequencing as a universal second-tier test in newborn screening
Genetics in Medicine (2021)
