
Abstract
Purpose
Albinism is a clinically and genetically heterogeneous condition. Despite analysis of the 20 known genes, ~30% patients remain unsolved. We aimed to identify new genes involved in albinism.
Methods
We sequenced a panel of genes with known or predicted involvement in melanogenesis in 230 unsolved albinism patients.
Results
We identified variants in the Dopachrome tautomerase (DCT) gene in two patients. One was compound heterozygous for a 14-bp deletion in exon 9 and c.118T>A p.(Cys40Ser). The second was homozygous for c.183C>G p.(Cys61Trp). Both patients had mild hair and skin hypopigmentation, and classical ocular features. CRISPR-Cas9 was used in C57BL/6J mice to create mutations identical to the missense variants carried by the patients, along with one loss-of-function indel. When bred to homozygosity the three mutations revealed hypopigmentation of the coat, milder for Cys40Ser compared with Cys61Trp or the frameshift mutation. Histological analysis identified significant hypopigmentation of the retinal pigmented epithelium (RPE) indicating that defective RPE melanogenesis could be associated with eye and vision defects. DCT loss of function in zebrafish embryos elicited hypopigmentation both in melanophores and RPE cells.
Conclusion
DCT is the gene for a new type of oculocutaneous albinism that we propose to name OCA8.
Similar content being viewed by others

INTRODUCTION
In humans, albinism defines a group of genetic diseases commonly subdivided into three main types: oculocutaneous albinism (OCA), ocular albinism (OA), and syndromic albinism (Hermansky–Pudlak syndrome [HPS] and Chediak–Higashi syndrome [CHS]).1,2 Generalized hypopigmentation of the skin and hair is highly variable among patients, ranging from total absence of melanin to undetectable defects in pigmentation. On the other hand, ophthalmological anomalies are more consistently detected and are therefore considered to be central in the definition of human albinism. These include iris and/or retinal hypopigmentation, nystagmus, foveal hypoplasia, chiasm misrouting of the optic nerves, and overall reduced visual acuity. Kruijt et al. described major and minor criteria and proposed that patients should display three major criteria or two major criteria and two minor criteria for the diagnosis of albinism to be ascertained.3
Albinism is also genetically heterogeneous since 20 genes are so far known to be involved (6 for OCA, 1 for OA, 1 for the closely related FHONDA, 11 for HPS, 1 for CHS).2,4,5,6 Despite a systematic search for both single-nucleotide variants and copy-number variants in 19 of these genes, 27% of patients remain without a molecular diagnosis.5
We and others have shown that over 50% of OCA patients with molecular diagnosis have pathogenic variants in TYR (OCA1) encoding tyrosinase while around 2–3% have a defective TYRP1 (OCA3) encoding tyrosinase-related 1 protein.1,5,7,8,9 Both enzymes are well known to control pigment synthesis.10 Of note, a third enzyme, dopachrome tautomerase, (DCT) acts downstream of TYR but upstream of TYRP1 on the DHICA melanin synthesis pathway11,12 making the corresponding gene (DCT/TYRP2) an obvious candidate for human albinism. However, pathogenic variants in DCT/TYRP2 have not yet been reported.
In the present study, we screened a series of 230 unsolved patients for pathogenic variants among a selection of candidate genes including DCT/TYRP2. We describe two patients with classical ocular albinism and mild hypopigmentation of the skin, hair, and eyes who both have potential pathogenic DCT/TYRP2 genotypes. We further validate these variants by analyzing the phenotypes of genetically modified mice lines and zebrafish embryos.
This work provides evidence that pathogenic variants in DCT/TYRP2 can cause oculocutaneous albinism in humans. We therefore propose that DCT/TYRP2 is now associated to a new OCA subgroup, OCA type 8 (OCA8).
MATERIALS AND METHODS
Ethics statement
This study was approved by the Comité de Protection des Personnes Sud-Ouest et Outre-Mer III. Informed consent was obtained from the patients and/or their parents in the case of minors before genetic analysis was performed. Authorization for publication, including the publication of photographs, was obtained from patient 1.
Sequencing of candidate genes
DNA was extracted from peripheral blood leukocytes using an automated procedure (Tecan EVO-ReliaPrep, Promega). Next-generation sequencing (NGS) was performed using the ionTorrent technology on a S5XL instrument (Life Technologies, Thermo Fisher Scientific, USA) (AmpliSeq panel) and bioinformatics analysis of variants were as previously described.5 DCT variants (NM_001129889.2) were confirmed by Sanger sequencing. See Supplementary Data for more details.
CRISPR modeling of Dct variants in the mouse
Mouse lines
All mouse work was carried out in compliance with United Kingdom Home Office regulations under a United Kingdom Home Office project license. To create the Dct118 A/A, Dct183 G/G, and Dct−/− alleles the CRISPR design site https://chopchop.cbu.uib.no/ (in the public domain) was used to design guides with default settings (g1 for Dct118 A/A and g2 for Dct183 G/G and Dct−/−, Supplementary Materials). Genetic engineering of CRISPR-Cas9 lines with each of the Cys substitutions was performed as already described13 and detailed in Supplementary Materials and Methods.
Direct spectrophotometry of Dct mice
Coat color of live mice, aged between 1 and 2 months, was measured by direct spectrophotometry using Konica Minolta CM-2600d, as already described14 and detailed in Supplementary Materials and Methods. Between five and nine animals were studied, depending on the strain.
Eye histology
Mice were sacrificed by cervical dislocation and eyes enucleated and processed as detailed in Supplementary Materials and Methods.
Randomization was not required, since comparison was between genotypes not treatments. Spectrometric measurements were carried out blind to genotype.
dct knockdown in Danio rerio
dct targeting
Zebrafish (D. rerio) were produced in accordance with the French Directive (Ministère de l’Agriculture) and in conformity with the European Communities Council Directive (2010/63/EU). The zebrafish homolog of DCT was identified by searching in the genome sequence database at http://www.ensembl.org/Danio_rerio taking into account the synteny with the human genome. Morpholinos (MOs) were designed by and obtained from GeneTools, LLC and embryo injection is detailed in Supplementary Materials and Methods.
Melanin assay was based on already described methods15 and detailed in Supplementary Materials and Methods.
Histological analysis was performed as described in Supplementary Materials and Methods.
RESULTS
Identification of DCT variants in patients with oculocutaneous albinism
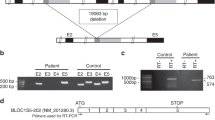
We recently described the analysis of 990 index cases with albinism and showed that a molecular diagnosis could be obtained after analysis of the 19 albinism genes known at that time in 72% of patients.5 Our cohort nowadays comprises more than 1500 patients, among whom the proportion of patients remaining without a molecular diagnosis is in the 25–30% range. Following the hypothesis that some of these patients may have pathogenic variants in genes other than the 19 already identified, we screened the genomes of 230 undiagnosed patients for variants in candidate genes selected for their known or predicted involvement in melanogenesis, including orthologues of mouse mutants with pigmentary defects (see Supplementary Table 1). We recently described the finding of BLOC1S5 variants in two patients with Hermansky–Pudlak syndrome (HPS11).6
Here, we identified homozygous or compound heterozygous variants of the DCT gene (NM_001129889.2), also called TYRP2, in two patients.
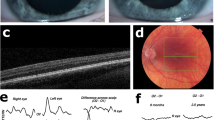
Patient 1, a 12-year-old girl born to unrelated parents of French origin, was diagnosed at the age of 3 years, and presented with moderate hypopigmentation of the skin and hair, congenital nystagmus, moderate foveal hypoplasia grade I,16 iris transillumination, and hypopigmentation of the retina (Fig. 1). Her visual acuity was 5/10 in both eyes. She harbored a single-nucleotide variant in exon 1 of DCT, c.118T>A p.(Cys40Ser) (ClinVar submission number SUB7603838), that was inherited from her heterozygous father, and a 14-bp deletion in exon 9, c.1406_1419del p.(Phe469*) (ClinVar submission number SUB7603850), inherited from her heterozygous mother. Patient 2 (age at diagnosis not known) is a woman aged 36 at the time of consultation who was born to consanguineous parents originating from Northern Africa. She had cream-colored skin, light brown hair, nystagmus, photophobia, iris transillumination, hypopigmentation of the retina, and reduced visual acuity (4/10 and 5/10). Morphology of the fovea was not investigated. Patient 2 was homozygous for variant c.183C>G p.(Cys61Trp) (ClinVar submission number SUB7603854) in DCT exon 1 (Fig. 1). Segregation analysis could not be completed and no further information on her phenotype could be gathered, as she was lost from contact after DNA sampling. All three variants were predicted to be deleterious by several prediction softwares (see “Materials and Methods”) taking into account parameters such as allele frequency in public genome databases, conservation, as well as effect on protein structure and physicochemical properties. All three variants are classified as likely pathogenic according to American College of Medical Genetics and Genomics criteria:17 c.118T>A p.(Cys40Ser) (PS3, PM2, PP3, PP4), c.1406_1419del p.(Phe469*) (PM2, PM4, PP3, PP4), c.183C>G p.(Cys61Trp) (PS3, PM2, PP3, PP4).

Photographs show the hypopigmentation of hair and skin of patient 1 (a, b). Bilateral foveal hypoplasia is seen in patient 1: (c) right (d) left eye), compared with a normal control (e) and a positive control (OCA1 patient) (f). Pedigree of patient 1 shows inheritance of the two variants from heterozygous parents (g). Pedigree of patient 2 is shown in (h). The patient’s consanguineous parents could not be tested because they were not available.
The c.1406_1419del p.(Phe469*) out-of-frame deletion present in patient 1 is predicted to result either in a truncated DCT protein lacking the transmembrane domain, which is necessary for anchoring the protein in the melanosomal membrane, or in messenger RNA (mRNA) nonsense-mediated decay.
Notably, both single-nucleotide variants found in the two patients, c.118T>A p.(Cys40Ser) and c.183C>G p.(Cys61Trp), affect highly conserved cysteine residues located in exon 1 of the gene and belonging to a Cys-rich domain that is part of an EGF-like domain with five potential disulfide bonds (Fig. 2).

In addition to Homo sapiens protein sequences of tyrosinase family including TYR, TYRP1, and TYRP2 (DCT), DCT protein sequences of Mus musculus, Gallus gallus, Xenopus tropicalis, and Danio rerio were aligned. Black shaded residues indicate more than 80% of identity whereas gray shaded residues indicate more than 80% of similarity. Black stars (*) highlight conserved cysteine residues. Black diamond indicates the position of the fortuitous out-of-frame 5-bp deletion in the knockout mouse model produced in the present study (p.Asn78*). Above the sequence alignment, the gray line indicates the EGF-like domain whereas the black dotted lines indicate the metal ion-binding domains. The black lined box represents the transmembrane domain. Black dots under the alignment indicate the previously reported slaty mutations in mouse (slt: R194Q; slt2J: P434L; sltLt: G486R). Numbering under alignment indicates the cysteine bounds.
These findings suggest that the three DCT variants identified are pathogenic and responsible for the albinism phenotype of the two patients described here. To further evaluate their pathogenicity, the two missense variants were introduced in mouse by CRISPR-Cas9 engineering.
CRISPR modeling of Dct variants in the mouse
In addition to the Dct mouse knockout line that was described by Guyonneau et al.,18 three different missense variants have been shown to be responsible for the grayish (slaty) color of mouse hair.12,19 As indicated in Fig. 2, none of these mutations are equivalent to the patients’ variants identified in this study. We therefore decided to genetically engineer CRISPR-Cas9 lines with each of the Cys substitutions.
Two lines were obtained with the exact equivalent changes to human p.(Cys40Ser) ([mouse c.118T>A p.[Cys40Ser]) and human p.(Cys61Trp) [mouse c.183C>G p.(Cys61Trp)]. Targeting of base pair 183 also led to a fortuitous out-of-frame 5-bp deletion (c.171_175del) in one founder that was brought to homozygosity as a loss-of-function control Dct−/− (frameshift results in premature stop codon at position 78, see Supplementary Data).
Mice heterozygous for any of the three mutations had a normally pigmented (black, nonagouti) fur. We crossed each line to produce homozygous mice that were grown to adulthood and all mice displayed dark gray hair, apparently identical to that seen in the previously described mutations of Dct (Fig. 3a). The color difference between mutant and control was much clearer in the ventral hair. The mouse ventrum is often paler than the dorsum, even in nonagouti mice such as these. Nonhairy skin (pinna and tail) of mutants are not obviously paler than control. To more precisely quantify the pigmentary changes we colorimetrically assayed the dorsal coat of the mutants in three-dimensional CIELAB color space, measuring along light–dark, blue–yellow, and red–green axes (Fig. 3b). All three lines were very different from the parental C57BL6/J mice. In addition, the Cys61Trp mice and the deletion mice were indistinguishable in all three dimensions. Interestingly, although not apparently different by eye, the mice homozygous for Cys40Ser were consistently less severely affected than the Cys61Trp and deletion mice in all three dimensions.

(a) Dorsal (upper panel) and ventral (lower panel) view of Dct mutant mice compared with C57Bl/6J controls. Male mice are shown, approximately 3 months old. (b) Three-dimensional CIELAB color measurement along light–dark, blue–yellow, and red–green axes as indicated for Cys40Ser, Cys61Trp, Dct−/−, and C57Bl/6J. For each measurement, the difference between any of the mutated strain and control was highly significant (t-test, p values < 10−6), no significant difference was found between Cys61Trp and Dct−/− whereas Cys40Ser was significantly less affected than Cys61Trp (especially in the red–green pigment contents, p value < 5.10−4). (c) Histology of 2-month-old retinas from Dct−/− and age-matched C57Bl/6J control mice. Fields were photographed at mid-distance between the optic nerve and ciliary body for both samples; scale bar 25 μm. One field is magnified to illustrate the significant difference in retinal pigment epithelium (RPE) pigmentation. Hematoxylin stained nuclei are clearly visible in Dct−/− (double arrow line) whereas nuclei are masked by intense pigmentation in control.
These observations in mice strongly suggest that the missense variants identified in both patients cause loss or partial loss of function of DCT and that this is the cause of their hypopigmentation of the hair and skin.
Dct knockout and slaty mice have hardly been investigated for ophthalmological anomalies. We conducted histological analysis of eyes from adult Dct−/− mice. Gross anatomy of the eye was not modified in these mice compared with wild-type (C57Bl/6J) age-matched controls although we cannot rule out that the outer nuclear layer was slightly thinner and disorganized in some Dct−/− retinas (not shown). Melanocytes in the choroidal layer appeared normally pigmented. However, careful examination of the outer retina evidenced that the retinal pigmented epithelium (RPE) in Dct−/− mice was significantly less pigmented than in wild-type retinas. As illustrated in Fig. 3c, dark melanin in the control eye completely masks the RPE cell membranes and contents whereas in Dct−/− eyes, pigmentation of the RPE is light enough that hematoxylin stained nuclei can be visualized in most of the RPE cells.
Altogether, these results show that when introduced in mice, Cys40Ser and Cys61Trp mutations result in hypopigmentation of the coat, the latter being undistinguishable from complete loss of function. Furthermore, RPE melanin content is reduced in Dct−/− mice, which may cause eye and vision anomalies in mice, and, in a similar way, in DCT patients.
dct knockdown in Danio rerio
Loss of function of dct was also investigated in zebrafish (D. rerio) embryos in which development and pigmentation of skin melanophores and RPE can be followed in vivo. Of note, only one DCT homolog is found in D. rerio genome and D. rerio protein sequences are highly conserved with 62% identity and more than 85% similarity (Fig. 2). The expression of dct was transiently knocked down using a morpholino (MO). Compared with embryos injected with MO-ctrl, MO-dct injected embryos did not show macroscopic phenotype disruption and no obvious disruption of skin melanophores pattern at neither 48 hpf nor 120 hpf (Fig. 4a, c). However, melanophores dorsally localized at the head level of 48 hpf MO-dct appeared lighter and wider than those of controls (Fig. 4a). Eye pigmentation was also slightly lighter in dct knocked-down embryos (Fig. 4a). To quantify a visible hypopigmentation, a whole-embryo melanin assay was performed, confirming a ≈30% significant decrease of total melanin in dct-knockdown embryos versus control embryos (Fig. 4b). Later in development at 120 hpf, MO-dct embryos still presented with a normal pattern of skin melanophores. However, their intracellular pigmentation was different from controls. In control embryos, pigmentation of melanophores appeared highly packed as expected whereas pigmentation in MO-dct embryos melanophores was consistently weaker and showed an abnormal stellar pattern (Fig. 4c). In addition to general pigmentation disruption, significant microphthalmia was noticed possibly due to off-target effect or toxicity, although other indicators for toxicity, e.g., shape and size of the embryos, were not significantly affected20 (Fig. 4d). Histological sectioning of 48 hpf embryos (Fig. 4a) evidenced eye developmental defects. In MO-Ctrl injected embryos, stratification of the neural retina into outer and inner layers was achieved by 48 hpf whereas at the same stage, MO-dct eyes still displayed only one homogeneous layer of neurons. The RPE in morphants was clearly hypopigmented, but in addition instead of the normal organization as a heavily pigmented cobblestoned single cell layer, the RPE showed disheveled cells and wavy contours.

(a) Representative lateral views of whole embryos (scale bar, 100 µm), dorsal views of heads (scale bar, 100 µm) and histological cross sections of eyes (scale bar, 20 µm) from representative 48 hpf zebrafish embryos injected with MO-ctrl or MO-dct. Bracket shows inner retina that has differentiated from outer retina in control but not in Mo-ctrl. (b) Melanin was quantified in 48 hpf zebrafish embryos injected with MO-ctrl or MO-dct. Means were calculated from the data collected from three independent experiments, Mann–Whitney test, *p < 0.05. (c) Representative lateral views of whole embryos at 120 hpf injected with MO-ctrl or MO-dct. Insets represent enlarged views of skin melanophores laterally localized in the trunk. (d) Embryo length (reported on left y-axis) and left eye area (right y-axis) were measured from one representative experiment on MO-ctrl group (n = 12) and MO-dct group (n = 28). Mann–Whitney test was applied for length data whereas unpaired t-test was performed for left area data. ns nonsignificant. **p < 0.01.
Altogether, these data are complementary to the observations made in mice, and confirm that loss of function of Dct impairs melanin metabolism in both melanophores and in RPE cells of early embryos in D. rerio with possible consequences on eye development and/or function.
DISCUSSION
We report the identification of pathogenic variants in the DCT gene in two unrelated patients with oculocutaneous albinism. Two of them are missense variants affecting highly conserved cysteine residues that belong to a Cys-rich domain (c.118T>A p.[Cys40Ser] and c.183C>G p.[Cys61Trp]). The third variant is a 14-bp out-of-frame deletion (c.1406_1419del p.[Phe469*]), a likely null allele. Both patients presented with a creamy skin and light brown hair, thus indicating that melanin is produced to a significant level. Visual acuity was moderately reduced (4/10–5/10) in both patients. The ocular phenotype included nystagmus, hypopigmentation of the retina and iris transilllumination. Foveal hypoplasia was of low grade in patient 1 and not investigated in patient 2.
Here we report DCT variants have been shown to cause oculocutaneous albinism in human. Dopachrome Tautomerase (DCT), formerly known as Tyrosinase-Related Protein 2, is one of three members of the tyrosinase protein family, along with TYR and TYRP1. All three are enzymes catalyzing reactions in the melanogenesis pathway, and are located within melanosomes, a pigment cell–specific organelle.10 In this series of reactions as previously reviewed,10 Tyrosinase (TYR) catalyzes the initial conversion of tyrosine to dihydroxyphenylalanine (DOPA) and DOPA to DOPAquinone, which spontaneously cyclizes to DOPAchrome. DCT catalyzes the tautomerization of DOPAchrome to dihydroxyindole carboxylic acid (DHICA). DOPAchrome can also spontaneously lose the carboxyl group to produce dihydroxyindole (DHI). The function of the third member of the family, Tyrosinase-related Protein 1 (TYRP1), is still not completely certain and may vary depending upon species and/or cell type. It probably acts, maybe in conjunction with TYR, to oxidize DHICA and DHI, by removing the two hydroxyl groups to the indole derivatives, which ultimately polymerize to form melanin on a protein matrix within the melanosome. The melanins formed from DHICA and DHI are eumelanins, which range in color from brown to black.
As TYR is the key limiting enzyme in melanogenesis, its absence results in complete lack of melanin, as seen in mutant mice and humans with OCA1. Absence of TYRP1 in mice results in the production of brown eumelanin, which appears to be less polymerized than black eumelanin. The human TYRP1 gene is altered in patients with OCA3. The effect on pigmentation, and ocular defects, are relatively mild with bright copper-red coloration of the skin and hair, both in African and European patients.5,21 Curiously, despite being upstream of TYRP1, the absence of DCT in mice results in a darker eumelanin than the brown eumelanin that lacks end-products generated in the presence of TYRP1. This is most likely due to DOPAchrome being converted to DHI, which produces a dark eumelanin. Absence of TYRP1 in presence of DCT means that DHICA, rather than its oxidized derivative, is polymerized to produce the brown eumelanin observed. A milder phenotype than the average OCA3 type is therefore expected in humans with a loss of DCT function.
The two missense variants identified in this study are located in the N terminal Cys-rich subdomain of DCT, which is strikingly conserved in its two paralogs TYR and TYRP1 and across species (Fig. 2). Localization of the ten cysteine residues (C1 to C10) in each enzyme is indicated in Table 1 as well as their participation in the five disulfide bonds that stabilize the domain.10
Note that C3 to C8 (involved in disulfide bonds 2, 3, 4) belong to the highly conserved EGF-like signature. According to the Human Gene Mutation Database, among 354 missense and nonsense pathogenic variants in TYR, substitutions in all of the C1 to C10 cysteine residues are found associated with OCA1. Of particular interest, C2 (patient 1 variant p.[Cys40Ser]) is substituted in two unrelated OCA1 Pakistani patients,22 and C5 (patient 2 variant p.[Cys61Trp]) is also substituted in OCA1 patients with three identified variants.23,24,25 Interestingly one of the OCA1 patients with a C2 substitution described by Jaworek et al.22 is homozygote for a Cys to Arg change and has a somewhat mild phenotype (fair blond hair and pinkish skin) compared with known complete loss of function. Moreover, the resulting TYR has a temperature-sensitive behavior likely due to slight protein misfolding. Whether this is due to loss of disulfide bond 1 and is therefore to be expected whatever amino acid is substituted at C1 or C2 would require further structural or functional investigations. Interestingly, the only allele involving a cysteine change of the Cys-rich domain in OCA3 patients affects C1 of TYRP1 and has also been associated with impairment of traffic to the melanosomes.26
Altogether, these data suggest that disruption of disulfide bond 1 in TYR and/or TYRP1 is likely to result in less severe and possibly temperature-sensitive misfolding of the protein. Ablation of bond 2 on the other hand may have a more detrimental effect on the structure of the whole enzyme as it is essential to stabilize the EGF-like domain.
Given the high degree of conservation of tyrosinase-related proteins between them and among species, we speculated that substitution of C2 and C5 in our DCT patients could have similar effects and investigated the corresponding phenotypes in mice. Supporting the relevance of this model for our comparative functional analysis, the two classical mutations of mouse in Tyr (albino) and Tyrp1 (brown) are also in conserved cysteines of the Cys-rich domain, in tyrosinase at C9, Cys103Ser,27,28 and in Tyrp1 at C8, Cys110Tyr.29 We show that mutation of Dct C2 in mice appears to be less severe than either C5 or the null frameshift that was produced. Although the difference is not readily detectable visually, the subtle variation is clearly seen when the fur is analyzed spectrophotometrically. It is tempting to attribute these differences to only a slight conformation change as in TYR and TYRP1. However, structural models of DCT/TYRP2 are speculative at this point as no crystallographic data have been so far generated.
Altogether these observations indicate a causal role of the C2 and C5 substitutions in our patients. The mild phenotype of patient 1 may be explained by a partial and possibly thermosensitive loss of function of DCT.
Previous studies18 have reported no effect of Dct mutations on mouse eye pigmentation. However, our close examination of mice with the null frameshift reveals a significant decrease in pigmentation of the RPE, although the choroid appears only mildly affected or not affected at all. How can this be related to some or all of the ophthalmic anomalies seen in the patients? Since our original demonstration that the RPE plays a key role in early development of the mammalian neural retina,30 a considerable amount of work has been performed, including in the albino Tyr−/− mouse, that sheds light on the primary cause for visual deficiency in OCA1 patients (for review see31). In particular it was shown that the lack of melanin and/or intermediates along the melanogenesis pathway leads to retinal developmental defects, especially in the neurogenesis of ganglion cells, and that defective guidance of their axons along the optic tracts results in optic chiasm misrouting and impairment of binocular vision (for review see32). Despite extensive investigation,33,34 it is still not clear which factors in the pigment synthesis pathway are involved and if they directly signal from the RPE to the neural retina or if they primarily affect RPE integrity. The fact that the RPE of both Dct−/− mice and zebrafish dct morphants is hypopigmented, together with typical ophthalmological defects diagnosed in our two DCT patients supports the hypothesis that the fully functional melanin pathway in the early RPE is instrumental to retinal neurogenesis.
The zebrafish embryo is a useful experimental model to study both skin and eye pigmentation as well as early eye development. Models for OCA1 and OCA3 have been described.35,36,37 Although dct expression pattern in zebrafish embryo has been used as a molecular marker of pigmented cells,38 its knockdown has not been previously described. Here we show that it results in hypopigmentation of both melanophores and RPE. In addition, instead of harboring their usual pigmentation pattern, melanophores of the skin resemble those of wild-type embryos responding to hormone-driven adaptation to surrounding darkness by melanin dispersion,36 suggesting that dct knockdown may impair melanin aggregation or that MO treatment has affected intracellular signaling. In the RPE, in addition to hypopigmentation, cell integrity was obviously affected. Such a pleiotropic effect has been described previously.39 Further investigations will aim at confirming and studying these changes at the cellular and molecular levels during development both in mouse and zebrafish embryos.
In conclusion, we report here evidence of DCT variants causing OCA in two unrelated patients. It is notable that both patients had mild hypopigmentation of the skin, hair, and eyes and moderate loss of visual acuity. Albinism is clinically heterogeneous, and DCT patients seem to fall in the mild range of the phenotypical spectrum. These mild phenotypes may be overlooked by clinicians and lead to underdiagnosis of albinism. As we recently discussed,40 we advocate that minimal signs, both dermatologic and ophthalmologic, sometimes of atypical presentation, should be systematically given consideration and lead to genetic investigations to establish a precise diagnosis. The DCT gene should now be included in albinism diagnostic gene panels. We propose that the corresponding form of OCA is called OCA8.
References
Grønskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007;2:43–51.
Montoliu L, Grønskov K, Wei AH, et al. Increasing the complexity: new genes and new types of albinism. Pigment Cell Melanoma Res. 2014;27:11–18.
Kruijt CC, de Wit GC, Bergen AA, Florijn RJ, Schalij-Delfos NE, van Genderen MM. The phenotypic spectrum of albinism. Ophthalmology. 2018;125:1953–1960.
Poulter JA, Al-Araimi M, Conte I, et al. Recessive mutations in SLC38a8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am J Hum Genet. 2013;93:1143–1150.
Lasseaux E, Plaisant C, Michaud V, et al. Molecular characterization of a series of 990 index patients with albinism. Pigment Cell Melanoma Res. 2018;31:466–474.
Pennamen P, Le L, Tingaud-Sequeira A, et al. BLOC1S5 pathogenic variants cause a new type of Hermansky–Pudlak syndrome. Genet Med. 2020. https://doi.org/10.1038/s41436-020-0867-5 [Epub ahead of print].
Gargiulo A, Testa F, Rossi S, et al. Molecular and clinical characterization of albinism in a large cohort of Italian patients. Invest Ophthalmol Vis Sci. 2011;52:1281–1289.
Hutton SM, Spritz RA. Comprehensive analysis of oculocutaneous albinism among non-Hispanic caucasians shows that OCA1 Is the most prevalent OCA type. J Invest Dermatol. 2008;128:2442–2450.
Zhong Z, Gu L, Zheng X, et al. Comprehensive analysis of spectral distribution of a large cohort of Chinese patients with non-syndromic oculocutaneous albinism facilitates genetic diagnosis. Pigment Cell Melanoma Res. 2019;32:672–686.
Lai X, Wichers HJ, Soler-Lopez M, Dijkstra BW. Structure and function of human tyrosinase and Tyrosinase-related proteins. Chemistry. 2018;24:47–55.
Murisier F, Beermann F. Genetics of pigment cells: lessons from the tyrosinase gene family. Histol Histopathol. 2006;21:567–578.
Budd PS, Jackson IJ. Structure of the mouse tyrosinase-related protein-2/dopachrome tautomerase (Tyrp2/Dct) gene and sequence of two novel slaty alleles. Genomics. 1995;29:35–43.
Cross SH, Mckie L, Keighren M, et al. Missense mutations in the human nanophthalmos gene TMEM98 cause retinal defects in the mouse. Invest Ophthalmol Vis Sci. 2019;60:2875–2887.
Mengel-From J, Wong TH, Morling N, Rees JL, Jackson IJ. Genetic determinants of hair and eye colours in the Scottish and Danish populations. BMC Genet. 2009;10:1–13.
Manga P, Sheyn D, Yang F, Sarangarajan R, Boissy RE. A role for tyrosinase-related protein 1 in 4-tert-butylphenol-induced toxicity in melanocytes: Implications for vitiligo. Am J Pathol. 2006;169:1652–1662.
Thomas MG, Kumar A, Mohammad S, et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography: a predictor of visual acuity? Ophthalmology. 2011;118:1653–1660.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Guyonneau L, Murisier F, Rossier A, Moulin A, Beermann F. Melanocytes and pigmentation are affected in dopachrome tautomerase knockout mice. Mol Cell Biol. 2004;24:3396–3403.
Jackson IJ, Chambers DM, Tsukamoto K, et al. A second tyrosinase-related protein, TRP-2, maps to and is mutated at the mouse slaty locus. EMBO J. 1992;11:527–535.
Bedell VM, Westcot SE, Ekker SC. Lessons from morpholino-based screening in zebrafish. Brief Funct Genomics. 2011;10:181–188.
Rooryck C, Roudaut C, Robine E, Müsebeck J, Arveiler B. Oculocutaneous albinism with TYRP1 gene mutations in a Caucasian patient. Pigment Cell Res. 2006;19:239–242.
Jaworek TJ, Kausar T, Bell SM, et al. Molecular genetic studies and delineation of the oculocutaneous albinism phenotype in the Pakistani population. Orphanet J Rare Dis. 2012;7:44–63.
King RA, Mentink MM, Oetting WS. Non-random distribution of missense mutations within the human tyrosinase gene in type I (tyrosinase-related) oculocutaneous albinism. Mol Biol Med. 1991;8:19–29.
Urtatiz O, Sanabria D, Lattig MC. Oculocutaneous albinism (OCA) in Colombia: first molecular screening of the TYR and OCA2 genes in South America. J Dermatol Sci. 2014;76:260–262.
Shahzad M, Yousaf S, Waryah YM, et al. Molecular outcomes, clinical consequences, and genetic diagnosis of Oculocutaneous Albinism in Pakistani population. Sci Rep. 2017;7:44185. https://doi.org/10.1038/srep44185.
Yamada M, Sakai K, Hayashi M, et al. Oculocutaneous albinism type 3: a Japanese girl with novel mutations in TYRP1 gene. J Dermatol Sci. 2011;64:217–222.
Yokoyama T, Silversides DW, Waymire KG, Kwon BS, Takeuchi T, Overbeek PA. Conserved cysteine to serine mutation in tyrosinase is responsible for the classical albino mutation in laboratory mice. Nucleic Acids Res. 1990;18:7293–7298.
Jackson IJ, Bennett DC. Identification of the albino mutation of mouse tyrosinase by analysis of an in vitro revertant. Proc Natl Acad Sci U S A. 1990;87:7010–7014.
Zdarsky E, Favor J, Jackson IJ. The molecular basis of brown, an old mouse mutation, and of an induced revertant to wild type. Genetics. 1990;126:443–449.
Raymond SM, Jackson IJ. The retinal pigmented epithelium is required for development and maintenance of the mouse neural retina. Curr Biol. 1995;5:1286–1295.
Mason C, Guillery R. Conversations with Ray Guillery on albinism: linking Siamese cat visual pathway connectivity to mouse retinal development. Eur J Neurosci. 2019;49:913–927.
Prieur DS, Rebsam A. Retinal axon guidance at the midline: chiasmatic misrouting and consequences. Dev Neurobiol. 2017;77:844–860.
Roffler-Tarlov S, Liu JH, Naumova EN, Bernal-Ayala MM, Mason CA. L-Dopa and the albino riddle: content of L-Dopa in the developing retina of pigmented and albino mice. PLoS One. 2013;8:e57184.
Lavado A, Jeffery G, Tovar V, De La Villa P, Montoliu L. Ectopic expression of tyrosine hydroxylase in the pigmented epithelium rescues the retinal abnormalities and visual function common in albinos in the absence of melanin. J Neurochem. 2006;96:1201–1211.
Richardson R, Tracey-White D, Webster A, Moosajee M. The zebrafish eye-a paradigm for investigating human ocular genetics. Eye. 2017;31:68–86.
Logan DW, Burn SF, Jackson IJ. Regulation of pigmentation in zebrafish melanophores. Pigment Cell Res. 2006;19:206–213.
Braasch I, Liedtke D, Volff JN, Schartl M. Pigmentary function and evolution of tyrp1 gene duplicates in fish. Pigment Cell Melanoma Res. 2009;22:839–850.
Lamason RL, Mohideen MAPK, Mest JR, et al. Genetics: SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005;310:1782–1785.
Gross JM, Perkins BD, Amsterdam A, et al. Identification of Zebrafish insertional mutants with defects in visual system development and function. Genetics. 2005;170:245–261.
Arveiler B, Michaud V, Lasseaux E. Albinism: an underdiagnosed condition. J Invest Dermatol. 2020;140:1449–1451.
Acknowledgements
The authors are grateful to the French Albinism Association Genespoir for financial support and for timeless action in favor of patients with albinism. I.G., L.M., M.K., and I.J.J. were funded by the MRC University Unit award to the MRC Human Genetics Unit, grant number MC_UU_00007/4. The authors also thank the patients for participating in this study and Angus Reid for assisting with colorimetric measurements.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Pennamen, P., Tingaud-Sequeira, A., Gazova, I. et al. Dopachrome tautomerase variants in patients with oculocutaneous albinism. Genet Med 23, 479–487 (2021). https://doi.org/10.1038/s41436-020-00997-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-00997-8
Keywords
This article is cited by
-
The experience of albinism in France: a qualitative study on dyads of parents and their adult child with albinism
BMC Medicine (2024)
-
Pigmentation and TYRP1 expression are mediated by zinc through the early secretory pathway-resident ZNT proteins
Communications Biology (2023)
-
Oculocutaneous albinism: the neurological, behavioral, and neuro-ophthalmological perspective
European Journal of Pediatrics (2023)
-
Integration of transcriptomics and metabolomics reveals amelanism mechanism of Oscar Astronotus ocellatus (Agassiz, 1831)
Hydrobiologia (2023)
-
Biallelic mutations in l-dopachrome tautomerase (DCT) cause infantile nystagmus and oculocutaneous albinism
Human Genetics (2021)
