
Abstract
Purpose
Detection of all major classes of genomic variants in a single test would decrease cost and increase the efficiency of genomic diagnostics. Genome sequencing (GS) has the potential to provide this level of comprehensive detection. We sought to demonstrate the utility of GS in the molecular diagnosis of 18 patients with clinically defined Alagille syndrome (ALGS), who had a negative or inconclusive result by standard-of-care testing.
Methods
We performed GS on 16 pathogenic variant-negative probands and two probands with inconclusive results (of 406 ALGS probands) and analyzed the data for sequence, copy-number, and structural variants in JAG1 and NOTCH2.
Results
GS identified four novel pathogenic alterations including a copy-neutral inversion, a partial deletion, and a promoter variant in JAG1, and a partial NOTCH2 deletion, for an additional diagnostic yield of 0.9%. Furthermore, GS resolved two complex rearrangements, resulting in identification of a pathogenic variant in 97.5% (n = 396/406) of patients after GS.
Conclusion
GS provided an increased diagnostic yield for individuals with clinically defined ALGS who had prior negative or incomplete genetic testing by other methods. Our results show that GS can detect all major classes of variants and has potential to become a single first-tier diagnostic test for Mendelian disorders.
Similar content being viewed by others


INTRODUCTION
Targeted applications of next-generation sequencing (NGS) tests, such as NGS panels, and screening of all protein-coding genes by exome sequencing (ES), are the current standard-of-care diagnostic tests for many suspected Mendelian disorders.1 While the use of targeted NGS tests has increased the overall diagnostic yield over the years, a molecular cause is not identified in roughly 70% of individuals1 who present for genomic testing. However, the diagnostic utility of NGS panels and ES varies based on the clinical indication, with some diseases exhibiting much higher yields than others.2,3 Regardless, there is a need to push beyond the current standard-of-care testing methods to increase diagnostic yield. Although ES is able to detect exonic sequence variants (single-nucleotide variants [SNVs] and insertion/deletions [indels]) with high confidence, ES is less equipped to comprehensively identify some of the major classes of genomic variation, namely copy-number and structural variants,1 which are typically detected by other approaches such as chromosomal microarray analysis (CMA), multiplex ligation-dependent probe amplification (MLPA), cytogenetic analysis, and fluorescence in situ hybridization (FISH). By design, ES does not usually identify deep intronic, promoter, and noncoding variants. Genome sequencing (GS) is poised to overcome many of the barriers faced by ES for several reasons. GS provides read coverage across both intronic and intergenic regions of the genome, enabling the comprehensive detection of all coding and noncoding genomic variants at nucleotide-level resolution, which greatly enhances clinical interpretation. In addition, polymerase chain reaction (PCR)–free protocols for GS eliminate amplification bias (a known confounder) to provide more uniform coverage compared with ES.
We sought to demonstrate the clinical utility of GS in identifying pathogenic variants in individuals with clinically defined Alagille syndrome (ALGS; OMIM 118450) using a cohort of 18 patients with previously negative or inconclusive testing. ALGS is an autosomal dominant disorder characterized by hepatic, cardiac, ocular, vertebral, renal, vascular, and facial involvement.4 It has long been recognized as a disease of Notch signaling deficiency, with 94.3% of individuals found to have a pathogenic variant in the Notch ligand, JAGGED1 (JAG1) and 2.5% of individuals found to have a pathogenic variant in the Notch receptor, NOTCH2.5 Standard-of-care testing to identify pathogenic variants typically employs a serial testing strategy that includes (1) sequencing JAG1 for SNVs and indels using genomic DNA; (2) performing deletion/duplication analysis of JAG1 using various strategies, but commonly MLPA and/or CMA; and (3) sequencing NOTCH2 for SNVs and indels using genomic DNA.6 Functional studies strongly suggest haploinsufficiency as a disease mechanism for JAG1 pathogenic variants, with a majority leading to early protein truncations,5 and a handful of studied missense variants leading to the translation of a nonfunctional or incorrectly trafficked protein product.5,7,8 The mechanism by which pathogenic NOTCH2 variants cause ALGS is less clear. No genes other than JAG1 and NOTCH2, including other Notch signaling genes, have been identified to cause ALGS. Thus, we hypothesized that individuals with clinically consistent ALGS and without a molecular diagnosis are likely to have a pathogenic variant within JAG1 or NOTCH2 that is undetectable by current screening methodologies. Using a previously identified cohort of well-characterized probands with clinically consistent ALGS, but with no confirmed JAG1 or NOTCH2 pathogenic variant, we aimed to assess the utility of GS in increasing the molecular diagnostic yield for this disease.
MATERIALS AND METHODS
Patient cohort
We have actively enrolled patients with suspected ALGS into our single-center ALGS research study (“Molecular Analysis of Alagille Syndrome”) at the Children’s Hospital of Philadelphia (CHOP) since 1992, amassing a convenience series of 446 probands, with participant referral occurring both within CHOP and worldwide through physician outreach to our study team. For this prospective study, we reviewed our database of 446 individuals with suspected ALGS and identified a cohort of 18 individuals enrolled between July 1997 and July 2014 with a clinical diagnosis of ALGS who had prior negative or inconclusive testing of sequence (via Sanger and/or NGS-based analysis) and copy-number variants (by MLPA and/or FISH) in JAG1, and sequence variants in NOTCH2 (Fig. 1 and Table S1).

Genome sequencing (GS) was performed on a cohort of 18 individuals that were identified in our study, Molecular Analysis of Alagille Syndrome (ALGS). Exclusion criteria and results of the study are indicated.
To establish our study cohort of 18 probands, we excluded those who did not meet the clinical diagnostic criteria for ALGS after medical record review, those with insufficient clinical information to support a clinical diagnosis of ALGS, those with no remaining sample for study, and those with low sample quality (Fig. 1). We also excluded individuals with a pathogenic variant in either JAG1 or NOTCH2 previously identified by standard-of-care testing. All 18 probands in the remaining group underwent chart review by a clinical team of two gastroenterologists and two geneticists at CHOP, and had at least three of five characteristic features of ALGS (Table S2). Sixteen of 18 probands had previously negative genomic testing and two had an inconclusive MLPA result for JAG1. These two were included to determine whether GS could fully resolve an apparently complex pathogenic variant. Family members of five probands (one quad, two trios, and two duos) were also chosen for GS based on sample availability.
Ethics statement
All probands and family members were enrolled and consented to participate in a research study approved by the Institutional Review Board at CHOP.
Standard-of-care testing
We employed a serial testing strategy for our standard-of-care testing that included a minimum of three genomic tests to assay for (1) small sequence variants such as SNVs and indels within the JAG1 coding region, including splice acceptor/donor variants; (2) full and partial gene deletion/duplication analysis of JAG1; and (3) SNVs and indels within the NOTCH2 coding region, including splice acceptor/donor variants. Previous studies have shown that these three testing strategies are capable of detecting up to ~97% of pathogenic variants in ALGS.5,6
Genome sequencing
DNA was extracted from either whole blood or lymphoblastoid cell lines using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). Short-read (2 × 150 bp) Illumina (Illumina, San Diego, CA) GS was performed at the Broad Institute Genomic Services (Boston, MA) using a PCR-free protocol at a targeted mean sequencing depth of 30×.9 BWA10-aligned CRAM files (hg38) produced by the GATK best practices workflow11 were obtained from the Broad Institute. Initial quality control steps included the estimation of coverage using the software tool indexcov,12 and the pairwise relatedness and sex-check using somalier.13
Variant calling and prioritization
SNVs and indels were called using the Strelka2 software14 and filtered using the genome intervals for JAG1 (hg38 chr20:10628605-10683078) and NOTCH2 (hg38 chr1:119911553-120069662) with 1 kb padding on either side to include variants in the promoter and untranslated regions. Annotation of SNVs/indels was performed using the software annovar.15 Genome-wide copy-number detection was performed using CNVnator16 and ERDS17 (mean read-depth using bins of length 1 kb) while structural variants were identified using manta18 with default settings. We filtered for copy-number and structural variants overlapping the above genomic intervals including JAG1/NOTCH2. All identified variants were manually inspected using the Integrative Genomics Viewer (IGV) visualization software (Broad Institute, Boston, MA). Rare SNVs and indels were filtered using a maximum minor allele frequency threshold of 0.1% in gnomAD v2.1 (https://gnomad.broadinstitute.org/)19 with the software slivar (https://github.com/brentp/slivar). Synonymous and intronic variants were annotated using the tool spliceAI.20 SpliceAI produces probability (DELTA score) for loss or gain of an acceptor or donor site. We used the recommended threshold of 0.5 to filter for cryptic splice acceptor/donor variants. We used genomic coordinates from ORegAnno database to filter for variants that fall within potential JAG1/NOTCH2 regulatory regions.
Variant confirmation
The deletion identified in proband 10, the inversion identified in proband 12, and the promoter variant identified in proband 11 were confirmed using standard droplet digital PCR (ddPCR) assays, which are described in the Supplementary Materials and Methods.
Complex rearrangements
Paired-end reads with abnormal insert sizes and soft-clipped reads that span the breakpoints were analyzed, and blat21 was used to map the soft-clipped reads across the breakpoint junctions to map the novel breakpoint junctions. Orientation of the read-pairs with abnormal insert sizes was used to infer inversions.
RESULTS
Of the 446 individuals in our database with both suspected and molecularly confirmed ALGS collected within our Molecular Analysis of ALGS research study at CHOP, we identified a cohort of 16 probands with prior negative standard-of-care testing and two probands with inconclusive MLPA results showing noncontiguous deletions who confidently met the clinical classification of ALGS (Fig. 1, Table S1). This cohort included ten males and eight females with a median age at enrollment of 5.7 years (range 3 months to 26 years) and with a highly diverse geographical distribution (Table 1). All 18 individuals presented with hepatic manifestations of ALGS, which included the presence of one or more of the following features: bile duct paucity, cholestasis, elevated liver enzymes, and cirrhosis (Table S2). Probands were also assessed for the presence of skeletal, cardiac, renal, ocular, and facial phenotypes as well as family history of ALGS (Table S2). All probands presented with at least three of these clinical features.
GS was performed in this ALGS cohort of 18 probands, specifically focusing on sequence, copy-number, and structural variants in JAG1 and NOTCH2, and resulted in the identification of a pathogenic variant in 6 of the 16 individuals with prior negative testing, including two deletions, an inversion, a promoter SNV, a JAG1 missense variant (c.401T>C; p.L134S), and a JAG1 frameshift variant (c.1978del; p.E660Rfs*83). GS further resolved the breakpoint architecture of both complex structural variants that were previously identified via MLPA (Table S2). Both the missense and frameshift variants identified in JAG1 were detectable by standard-of-care sequencing, and subsequent review of the original raw sequencing data showed that both of these variants were present, but were missed by the analyst. Of the remaining four novel variants that were identified by GS, three were within JAG1 and one deletion was within NOTCH2. There were no cryptic splice site or potential regulatory variants identified by spliceAI (DELTA score ≥0.5) and ORegAnno database, respectively.
The pathogenic variants resolved by GS included an inversion, a promoter variant, an exon 1 deletion in JAG1, and a deletion in NOTCH2. In proband 12, a 672-kb copy-neutral inversion involving the first three exons of JAG1 (chr20:10,663,195–11,342,633) was identified (Fig. 2, Fig. S1), which was inherited from his clinically affected father (Table S2). The 5’ end of the inversion mapped to intron 3 of JAG1 and the 3’ end mapped outside of the JAG1 gene, within a gene desert. Gene expression analysis of JAG1 using ddPCR confirmed that JAG1 expression was reduced in both proband 12 and his father (proband 12-F), suggestive of JAG1 haploinsufficiency (Fig. 3a). In proband 11, a novel SNV (c.-100G>A; chr20:10,673,630), of unknown inheritance, that is absent from public genomic variant databases (ExAC and gnomAD; https://gnomad.broadinstitute.org/)19 was identified in the promoter region of JAG1 (Fig. S2). Gene expression analysis of JAG1 using ddPCR confirmed that JAG1 expression was reduced in proband 11, suggestive of JAG1 haploinsufficiency (Fig. 3a). The two deletion variants included a maternally inherited (from an affected mother) 606-bp deletion involving exon 1 of JAG1 (chr20:10,673,044–10,673,649), identified in proband 15 (Fig. S3), and a de novo 5.9-kb deletion involving exons 31–34 of NOTCH2 (chr1:119,913,673–119,919,578), identified in proband 10 (Fig. 3b, Fig. S4).

The reference genome (upper structure) depicts the 679-kb inverted region, encompassing JAG1 exons 1–3, bounded by dashed lines. The breakpoints extend from intron 3 to a gene desert upstream of the JAG1 promoter. The rearranged structure is shown below. Paired-end reads with abnormal insert size and orientation were used to infer the approximate boundaries of the inversion and soft-clipped reads at the ends of the inversion were used to precisely map the breakpoints at nucleotide-level resolution.

(a) Droplet digital polymerase chain reaction (ddPCR) performed on complementary DNA (cDNA) made from RNA extracted from lymphoblastoid cell lines of affected individuals showing reduced JAG1 gene expression in the individual with the promoter variant (proband 11) as well as the individual with the inversion (proband 12) and his affected father (proband 12-F), who also has the inversion. Parental samples were not available for proband 11. The negative control is the average of four unaffected individuals with no pathogenic JAG1 variant. An individual with a pathogenic frameshift variant (c.2122_2125del) that is predicted to truncate the JAG1 protein was included as a positive control. Two separate primer/probe sets were used for confirmation, one designed in exon 1 and one designed to cross exons 25–26. Values for all samples were normalized to the internal control, TBP. Error bars for the negative control are plotted as standard deviation. (b) ddPCR showing NOTCH2 copy number in proband 10 and her unaffected parents.
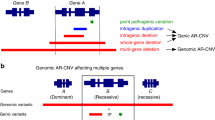
Additionally, we resolved the complex structural variants in the two individuals (probands 8 and 14, both of unknown inheritance) with prior MLPA results that were suggestive of a complex rearrangement involving multiple breakpoints and required more precise characterization (Fig. 4a). In proband 8, we confirmed the presence of the two previously identified noncontiguous deletions, involving exon 3 (9 kb) and exons 9–26 (23 kb), as well as a third deletion distal to the exon 3 deletion (4 kb), which was not known prior to GS (Fig. 4b, c and Fig. S5). Analysis of the read-pairs with abnormal insert sizes and orientation further revealed an inversion between the two intragenic deletions within JAG1 (Fig. 4b, c and Fig. S5). CNVnator identified all three deletions (22.9 kb, 9 kb, and 4.3 kb) while ERDS identified two of the three (9 kb and 4.3 kb). Manta identified the inversion with precise breakpoints with an 18-bp insertion at the distal breakpoint (chr20:10690781) (Table S3).

(a) JAG1 multiplex ligation-dependent probe amplification (MLPA) results for probands 8 and 14. Probe ratio is plotted for each exon. A threshold below 0.75 was used to classify losses and above 1.25 was used to classify gains. Circles represent copy-neutral ratios while squares represent deletions. The SALSA MLPA probemix (P184-C3 JAG1) was purchased from MRC Holland (Amsterdam, Netherlands) and details, including quantification, normalization, and controls can be found through this link: https://www.mrcholland.com/products/18527/Product%20description%20P184-C3-0317%20JAG1-v12.pdf. (b) Schematic of the genomic structure of JAG1 for proband 8, who had noncontiguous deletions of exon 3 and exons 9–26 demonstrated by MLPA, and proband 14, who had noncontiguous deletions of exon 9 and exons 11–12. Dashed lines denote the genomic coordinates (hg38) of the breakpoints and red bars indicate the deleted regions, which are interspersed with nondeleted portions of the gene (shown in gray). (c) Schematic of the genomic rearrangement for probands 8 and 14.
Similarly, analysis of GS data from proband 14 revealed a comparatively simpler JAG1 rearrangement but with smaller segments of DNA including an intragenic inversion involving exon 10 (246 bp) between two 1-kb deleted segments (exon 9 and exons 11–12) (Fig. 4b, c and Fig. S6). CNVnator identified a single contiguous 2.3-kb deletion and missed the normal region (246 bp) situated between the two 1-kb deletions. Manta identified two pairs of breakends and did not classify the breakends into a variant type (e.g., deletion) (Table S3).
DISCUSSION
Molecular diagnosis of ALGS can be accomplished by screening for pathogenic variants in JAG1 or NOTCH2 using standard techniques for sequencing and copy-number analysis, with a diagnostic rate of ~97%.5,6 In this study, we used GS on 18 patients, in whom pathogenic variants were not identified by standard methods, drawn from a larger patient cohort of 406 individuals. Of these 18 probands, 2 were found to have a pathogenic variant that was missed by Sanger sequencing, 2 had breakpoint mapping of complex rearrangements involving JAG1 to a resolution that was not attainable by MLPA, and 4 were found to have novel variants that could not be detected by previous testing methods. Therefore, 8/18 patients with no confirmed pathogenic variant identified by standard-of-care testing were resolved via GS, and 4 of these individuals were found to have a variant that would only be detectable through GS.
Each of the four novel pathogenic variants identified emphasizes a different diagnostic advantage of GS compared with ES, highlighting its diverse and striking clinical utility. Proband 15 was found to have a JAG1 exon 1 deletion, despite having a previously normal MLPA result. MLPA assays are limited to detect copy-number variants inside of a region bound by two probes. After reviewing the original MLPA data, we found that the distal breakpoint of this deletion fell 10 bp outside of the second exon 1 probe in the MLPA design. This highlights a limitation in MLPA testing and underscores the possibility of false negatives when utilizing this technology.
Proband 10 was found to have a deletion across exons 31–34 in the NOTCH2 gene. Copy-number variants in the NOTCH2 gene have not been previously reported in ALGS patients, and therefore copy-number analysis for NOTCH2 has not been recommended for standard-of-care testing.5 The pathomechanism of NOTCH2 variants is less clear than that for JAG1 variants, particularly since the majority of NOTCH2 variants are missense rather than protein-truncating.5 Truncating variants in the terminal exon (exon 34) of NOTCH2 have been implicated in Hajdu–Cheney syndrome, characterized by focal bone destruction and osteoporosis, along with other features (OMIM 102500). Hajdu–Cheney associated NOTCH2 pathogenic variants have been shown to escape nonsense-mediated decay and lead to gain-of-function protein products,22 a pathomechanism that is distinct from those proposed for NOTCH2 variants in ALGS. Minimal functional evidence from ALGS NOTCH2 variants is unable to confirm haploinsufficiency as a singular disease mechanism, and indeed a study examining the functional effect of a handful of NOTCH2 variants found that despite all of them displaying defective Notch signaling, one of the nonsense variants studied was shown to escape nonsense-mediated messenger RNA (mRNA) decay.23 Thus, the pathomechanism of NOTCH2 variants in ALGS appears to be varied, possibly including haploinsufficiency as well as other mechanisms. The phenotype of proband 10 in our cohort is remarkably different compared with Hajdu–Cheney syndrome and includes cholestasis, peripheral pulmonic stenosis, posterior embryotoxon, and classic ALGS facies. While we confirm the reduced copy number of NOTCH2 in proband 10, more functional evidence may be required to substantiate haploinsufficiency as the pathomechanism. Although we predict that NOTCH2 copy-number variants are a rare cause of ALGS, our results suggest that current testing guidelines should be reconsidered to include copy-number analysis for NOTCH2.
Our finding of a JAG1 promoter variant in proband 11, and the subsequent confirmation of reduced JAG1 gene expression in this proband, is particularly interesting as it provides new evidence that variants in JAG1 regulatory regions are capable of causing ALGS. We have previously suggested that ALGS patients who were not found to have an identified variant in JAG1 or NOTCH2 by conventional testing methodologies were likely to have variants in regulatory regions,5 and finding this promoter variant opens up the field to more exploratory research on JAG1 gene regulation, promising the potential to find disease-causing variants outside of protein-coding regions.
Lastly, the identification of an inversion involving the first three exons of JAG1 highlights the advantage of GS when it comes to detecting copy-neutral genomic rearrangements, which are missed by chromosomal single-nucleotide polymorphism (SNP) arrays. Moreover, the intronic position of one breakpoint requires GS technology, which provides equal coverage across coding and noncoding regions, rather than ES testing, which would fail to identify the intronic breakpoint.
GS also resolved previously unknown complex rearrangements in probands 8 and 14. We used two read-depth based copy-number variant callers (CNVnator and ERDS) and one split-read caller (manta) to identify these structural variants. CNVnator was the most sensitive among the read-depth callers finding all deletions (n = 5/5) while ERDS failed to detect deletions smaller than 2 kb. In proband 14, there were two deletions (918 bp and 1.2 kb) with a small 246-bp region of two-copy DNA between them. CNVnator failed to recognize the 246-bp normal region between the two deletions, most likely due to the fact that the size of the normal region is lower than the threshold used for binning the read-depth (1000 bp). Manta was able to identify the inversions overlapping these breakpoints. Read-depth callers provided approximate breakpoints while manta provided exact breakpoints involved in both structural variants. Thus, it is important to use a combination of read-depth and split-read-based callers to characterize complex rearrangements using GS data along with manual work to reconstruct the entire complex rearrangement.
In addition, we further identified two JAG1 variants that were missed by conventional Sanger sequencing. The frameshift variant (c.1978del; p.E660Rfs*83) has not previously been reported, but is expected to be pathogenic since it results in early protein truncation. The missense variant (c.401T>C; p.L134S) was previously reported in another individual within our ALGS cohort.5 Upon review of the original sequencing data, both of these variants were found to be present, and thus were missed by the original study, highlighting an analytical limitation of manual sequencing review.
Our study was limited to include only those individuals that had an available sample and that met a very conservative clinical diagnostic requirement, as determined by a team of two gastroenterologists and two geneticists. The identification of JAG1 and NOTCH2 pathogenic variants in individuals with mild ALGS, or who have less than three clinical symptoms, has been documented.24 In our testing scheme, these individuals were excluded from our GS analysis. Although this was a limitation of our study, we felt it was necessary to apply the most stringent clinical diagnostic guidelines to evaluate GS as a genomic tool in a population that was most likely to truly have ALGS. A second potential limitation for our study is that GS for all of the identified genetic variants in addition to their orthogonal variant confirmation utilize DNA/RNA from lymphoblastoid cell lines rather than primary tissue. However, the use of lymphoblastoid cell lines in the medical genetics literature is well established, and has had a very high success rate. While the possibility that somatic variants may arise during cell culture exists, the reported somatic variant rate is quite low (0.3%).25
Although we hypothesize that the remaining ten individuals who were not found to have a pathogenic variant identified may have variants in as-yet unidentified regulatory regions of, or cryptic splice variants in, JAG1 or NOTCH2, or were missed by the current bioinformatics methods, there is the possibility that some of these individuals may have a different disorder. Clinical phenotypes can emerge over time, and it is possible that new information may point to other molecular diagnoses in these patients.26,27 However, we attempted to minimize the likelihood of this outcome by choosing individuals with phenotypic features that were highly characteristic for ALGS. In the future, total mRNA sequencing of these individuals might help identify pathogenic alternatively spliced variants in JAG1/NOTCH2 missed by DNA sequencing.
Prior to GS, our diagnostic yield using standard-of-care testing for our clinically consistent ALGS probands within our Molecular Analysis of ALGS study was 96.6% (JAG1 n = 382/406, 94.1%; NOTCH2 n = 10/406, 2.5%; pathogenic variant negative n = 14/406, 3.4%). We report an additional diagnostic yield of 0.9% after applying GS to our testing strategy (JAG1 n = 385/406, 94.8%; NOTCH2 n = 11/406, 2.7%; pathogenic variant negative n = 10/406, 2.5%). We excluded both samples with complex rearrangements previously identified by MLPA (probands 8 and 14) and both samples with SNVs that were missed by Sanger sequencing (probands 17 and 18) from our increase in diagnostic yield since standard-of-care would be diagnostic for these four individuals.
Major deterrents to the utilization of GS as a first-tier genomic test include the higher sequencing costs and the burden of data analysis. By using a “genome slice” and analyzing only the two known disease genes known to cause ALGS, we significantly reduced the burden of data analysis yet we are able to detect all previously reported JAG1 and NOTCH2 pathogenic variants in ALGS as well as novel structural variants, intronic variants, promoter variants, and regulatory variants, with the capability to reflex back to the whole genome if necessary. Current diagnostics for ALGS involves sequential testing of at least three tests, and with our identification of a novel copy-number variant within the NOTCH2 gene, a fourth test involving copy-number evaluation of NOTCH2 should be added to this testing schema. The variety of variants identified in the cohort we describe here highlights the ability of GS to detect all major classes of variants, allowing for single-test diagnostics rather than serial testing strategies. As GS becomes more available as a clinical testing option, it is reasonable to recommend that it replace NGS panels and deletion/duplication analysis as a first-tier testing strategy for ALGS. A reduction in the time and costs associated with multiple tests may prove to be advantageous over the challenges in implementing GS as a first-tier diagnostic test for ALGS, and a similar advantage may be found in employing this testing strategy with other monogenic or oligogenic Mendelian disorders that require focused GS data analysis of only one or a few genes.
Data Availability
The GS datasets generated and analyzed during the current study are not publicly available as they are patient samples with unique mutations that are impossible to guarantee anonymity for.
References
Abou Tayoun AN, Krock B, Spinner NB. Sequencing-based diagnostics for pediatric genetic diseases: progress and potential. Expert Rev Mol Diagn. 2016;16:987–999.
Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18:696–704.
Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511.
Mitchell E, Gilbert M, Loomes KM. Alagille syndrome. Clin Liver Dis. 2018;22:625–641.
Gilbert MA, Bauer RC, Rajagopalan R, et al. Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Hum Mutat. 2019;40:2197–2220.
Gilbert MA, Spinner NB. Genetics of Alagille syndrome. In: Kamath BM, Loomes KM, editors. Alagille syndrome: pathogenesis and clinical management. Cham, Switzerland: Springer; 2018. p. 33–48.
Morrissette JD, Colliton RP, Spinner NB. Defective intracellular transport and processing of JAG1 missense mutations in Alagille syndrome. Hum Mol Genet. 2001;10:405–413.
Tada M, Itoh S, Ishii-Watabe A, Suzuki T, Kawasaki N. Functional analysis of the Notch ligand Jagged1 missense mutant proteins underlying Alagille syndrome. FEBS J. 2012;279:2096–2107.
Myles RB. Familial short oesophagus. Br J Radiol. 1939;12:645–647.
Cornell University. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. https://arxiv.org/abs/1303.3997. Accessed 16 October 2020.
Van der Auwera GA, Carneiro MO, Hartl C. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.10.33.
Pedersen BS, Collins RL, Talkowski ME, Quinlan AR. Indexcov: fast coverage quality control for whole-genome sequencing. Gigascience. 2017;6:1–6.
Pedersen BS, Bhetariya PJ, Brown J, et al. Somalier: rapid relatedness estimation for cancer and germline studies using efficient genome sketches. Genome Med. 2020;12:62.
Kim S, Scheffler K, Halpern AL, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018;15:591–594.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–1566.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984.
Zhu M, Need AC, Han Y, et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet. 2012;91:408–421.
Chen X, Schulz-Trieglaff O, Shaw R, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–1222.
Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443.
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–548. e524
Kent WJ. BLAT-the BLAST-like alignment tool. Genome Res. 2002;12:656–664.
Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. Semin Cell Dev Biol. 2012;23:450–457.
Kamath BM, Bauer RC, Loomes KM, et al. NOTCH2 mutations in Alagille syndrome. J Med Genet. 2012;49:138–144.
Guegan K, Stals K, Day M, Turnpenny P, Ellard S. JAG1 mutations are found in approximately one third of patients presenting with only one or two clinical features of Alagille syndrome. Clin Genet. 2012;82:33–40.
Sie L, Looneg S, Tan EK. Utility of lymphoblastoid cell lines. J Neurosci Res. 2009;87:1953–1959.
Ansari M, Poke G, Ferry Q, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J Med Genet. 2014;51:659–668.
Rexach J, Lee H, Martinez-Agosto JA, Nemeth AH, Fogel BL. Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurol. 2019;18:492–503.
Acknowledgements
This work was funded by the Fred and Suzanne Biesecker Pediatric Liver Center at the Children’s Hospital of Philadelphia (N.B.S., K.M.L., D.A.P) and by R01-HG009708 (R.R. and L.K.C.) and R01-DK081702-05 (N.B.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Co-First Authorship: Ramakrishnan Rajagopalan, Melissa A. Gilbert.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rajagopalan, R., Gilbert, M.A., McEldrew, D.A. et al. Genome sequencing increases diagnostic yield in clinically diagnosed Alagille syndrome patients with previously negative test results. Genet Med 23, 323–330 (2021). https://doi.org/10.1038/s41436-020-00989-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-00989-8
Keywords
This article is cited by
-
Molecular genetics of Parkinson’s disease: Contributions and global trends
Journal of Human Genetics (2023)
-
Genomic frontiers in congenital heart disease
Nature Reviews Cardiology (2022)
-
Best practices for the interpretation and reporting of clinical whole genome sequencing
npj Genomic Medicine (2022)
