
Abstract
Purpose
Pathogenic variants in the X-linked gene NEXMIF (previously KIAA2022) are associated with intellectual disability (ID), autism spectrum disorder, and epilepsy. We aimed to delineate the female and male phenotypic spectrum of NEXMIF encephalopathy.
Methods
Through an international collaboration, we analyzed the phenotypes and genotypes of 87 patients with NEXMIF encephalopathy.
Results
Sixty-three females and 24 males (46 new patients) with NEXMIF encephalopathy were studied, with 30 novel variants. Phenotypic features included developmental delay/ID in 86/87 (99%), seizures in 71/86 (83%) and multiple comorbidities. Generalized seizures predominated including myoclonic seizures and absence seizures (both 46/70, 66%), absence with eyelid myoclonia (17/70, 24%), and atonic seizures (30/70, 43%). Males had more severe developmental impairment; females had epilepsy more frequently, and varied from unaffected to severely affected. All NEXMIF pathogenic variants led to a premature stop codon or were deleterious structural variants. Most arose de novo, although X-linked segregation occurred for both sexes. Somatic mosaicism occurred in two males and a family with suspected parental mosaicism.
Conclusion
NEXMIF encephalopathy is an X-linked, generalized developmental and epileptic encephalopathy characterized by myoclonic–atonic epilepsy overlapping with eyelid myoclonia with absence. Some patients have developmental encephalopathy without epilepsy. Males have more severe developmental impairment. NEXMIF encephalopathy arises due to loss-of-function variants.
Similar content being viewed by others
INTRODUCTION
The developmental and epileptic encephalopathies (DEEs) are the most severe group of epilepsies in which frequent epileptic activity, in addition to the underlying etiology, contributes to developmental impairment, with onset typically in infancy or childhood.1 At least 50% of the DEEs have a genetic cause,2 and there is significant etiological overlap with other neurodevelopmental disorders such as intellectual disability (ID) and autism spectrum disorder (ASD).3 A subgroup of neurodevelopmental disorders are X-linked where phenotypic expression may vary markedly between males and females. e.g.:4,5 X-chromosome inactivation (XCI) is the epigenetic process where transcription of one of the X-chromosomes in female cells is silenced to balance gene dosage. This usually occurs in a random manner leading to cellular mosaicism. In females carrying a heterozygous X-chromosome pathogenic variant, skewing of XCI toward the wild type or mutant allele may protect them from, or predispose them to, disease.6,7
The Neurite EXtension and MIgration Factor (NEXMIF), previously called KIAA2022 (OMIM 300524), is an X-linked gene that is thought to play an important role in early brain development.8,9,10,11 Pathogenic NEXMIF variants were first identified in males with nonsyndromic X-linked ID with poor or absent speech, subtle dysmorphic features, and sometimes epilepsy.12,13 Heterozygous females in these families were mostly unaffected. Subsequently, a few affected females have been described14,15,16 and, in 2016, a series of 14 affected heterozygous females was reported.17 Females typically had generalized epilepsy and less severe intellectual impairment than males. The authors suggested that phenotypic variation between females may be attributable to XCI.
Here, we aimed to systematically evaluate the phenotypic spectrum of NEXMIF encephalopathy focusing on the epileptology and to study genotype–phenotype correlations in males and females in a large international cohort of patients.
MATERIALS AND METHODS
Patient ascertainment and phenotyping
Patients with NEXMIF encephalopathy were recruited through the Epilepsy Genetics Research program at the University of Melbourne and a NEXMIF European collaboration. Epilepsy, developmental, and medical histories were collected, with review of medical files, electroencephalogram (EEG) and magnetic resonance imaging (MRI) studies, and administration of an epilepsy questionnaire where possible. Seizures were classified according to the 2017 International League Against Epilepsy classification18 and, where possible, a specific epilepsy syndrome diagnosis was made.1,19 Video EEG recordings and MRI images were reviewed by an electrophysiologist (EG) and pediatric neuroradiologist (SM), where available. Clinical photographs (17 new, 1 published without a picture,20 11 published cases12,13,15,16,21,22) were reviewed and analyzed through the program Face2Gene (see below). Unaffected heterozygous females were not included in the data.
Genotyping and X-inactivation
NEXMIF variants in new patients were identified by collaborating research and diagnostic laboratories. Parental and family segregation studies were performed where possible. Pathogenicity of variants was assessed according to the American College of Medical Genetics and Genomics guidelines.23 Of 52 unreported patients, 46 carried pathogenic or likely pathogenic variants (43 singletons; 3 cases from one family) and 6 patients from 2 families carried variants of uncertain significance (VUS) reviewed separately. Due to the uncertain nature of both variants, we did not classify these individuals as having NEXMIF encephalopathy and did not include them in our analysis. All variants were absent in the Genome Aggregation Database (gnomAD).24 NEXMIF transcript NM_001008537.3 was used for coding variant nomenclature (Supplementary Table S1). Where possible, XCI testing in affected females was performed in blood in the diagnostic and research laboratories that identified the variant. X-inactivation was interpreted as skewed when ratios were ≥75:25.17
Literature review
A PubMed search was performed with the search terms “KIAA2022” or “NEXMIF” (search ended 10 April 2020). We selected all publications in English reporting (possibly) pathogenic NEXMIF variants, and identified 14 studies, reporting 41 patients. For 38 patients, clinical and genetic data were available for systematic review.12,13,14,15,16,17,21,22,25,26,27,28,29,30 For three reported patients, we collected additional clinical information.26,29 NEXMIF pathogenic variants reported in large data set studies with minimal or no clinical information were not included.
Automated facial analysis
Frontal facial 2D photographs of 27 individuals with NEXMIF encephalopathy were analyzed using the Face2Gene Research application (FDNA Inc., Boston, MA).31 This NEXMIF cohort was compared with 27 controls and 27 patients with SYNGAP1 encephalopathy (Supplementary methods). Patients with SYNGAP1 encephalopathy were chosen as their epileptology is similar to NEXMIF encephalopathy. Cohorts were matched for ethnicity, gender, and age.
Statistical analysis
All patients with pathogenic or likely pathogenic NEXMIF variants were included in our analyses. Only the index for each family was included in statistical analyses comparing males and females. Differences between males and females were calculated with a Fisher’s exact test for binary variables or a Mann–Whitney U test for continuous and ordinal variables. Statistical analyses were performed using SPSS statistics software (IBM SPSS Statistics 26). All reported tests were performed two-tailed with an alpha level for significance of p < 0.05.
Ethics statement
Written informed consent for participation/publication was obtained from all participants or their parents or legal guardians in the case of minors or adults with ID. Separate consent forms for publication of pictures/videos were obtained when applicable. This study was approved by the Human Research Ethics Committees of Austin Health, Australia and Western Zealand, Denmark.
RESULTS
Our cohort comprised 87 patients (78 families) with NEXMIF encephalopathy including 46 new patients, and 3 published patients with additional information26,29 (phenotypic data in Supplementary Table S2) and data from 38 published patients (Supplementary Table S3).12,13,14,15,16,17,21,22,25,27,28,30 The cohort included 24 males and 63 females; median age at study was 10 years (range 1–53 years). As we did not have complete information on all patients, denominators indicate the number in whom information on the clinical feature addressed was available.
Neurodevelopment, autism spectrum disorder, and behavioral features
All 24 males had developmental delay, observed prior to 1 year of age in 15/18 (83%) (Table 1). Median age of walking was 34 months (range 14 months–6 years). Two boys could not walk at 3 years and one lost the ability to walk 6 months after starting walking.13 Seven of 12 (58%) males older than 3 years were nonverbal, 3 (25%) spoke single words, and 2 (17%) spoke in simple sentences. Regression or stagnation in development occurred in 7/10 males (70%) at median age 22 months (range 5–40 months) and, in 5, coincided with seizure onset or increased seizure frequency. Seventeen of 23 (74%) males had severe to profound ID; 6/23 (26%) had mild to moderate ID.
Females with NEXMIF encephalopathy showed broader variation in developmental and cognitive profile. In comparison with males, onset of developmental delay was recognized later (<1 years in only 17/47, 36%, p = 0.006) with median age of walking significantly earlier at 18 months (range 11 months–4 years, p < 0.001). More females reached a higher level of expressive language, with 38/52 (73%) females older than 3 years speaking simple sentences (p < 0.001). Regression or stagnation in development occurred in 14/44 (32%) females, coinciding with seizure onset or increased seizure frequency in 5/9 females on whom this information was available. Accordingly, levels of ID ranged from mild to moderate in 41/61 (67%), to severe or profound ID in 19/61 (31%), the inverse pattern to the male cognitive profile (p < 0.001). Of note, there was 1/61 (2%) females who had generalized epilepsy without ID.17 Only affected females were included in our analysis. Healthy heterozygous females were described in several published X-linked families with affected males only12,13,25 and also found in our families (Supplementary Table S2).
In 22/54 (41%) patients with seizures on whom information was available, developmental delay preceded seizure onset. Moreover, 15/86 (17%) patients (8/23 [35%] males, 7/63 [11%] females) had developmental delay/ID without seizures.
Although ASD or autistic features were more frequently reported in males (18/24, 75%) than females (28/61, 46%), this difference did not reach statistical significance (p = 0.057) when including index patients only. In 60/85 (70%) patients, behavioral problems occurred, most frequently hyperactivity (25/85, 29%), often with attention deficit disorder (20/85, 24%), aggression (20/85, 24%), and temper tantrums (10/85, 12%). Stereotypies were observed in 13/85 (15%) patients.
Seizure semiology
Seizures were present in 71/86 (83%) patients, less frequently in males (15/23, 65%) than females (56/63, 89%, p = 0.005). One reported male had electrical seizures without clinical correlate and was not included in our analysis.25 Seizure onset age did not significantly differ between males (median 15 months, range 1 month–14 years) and females (median 19 months, range 1 month–17 years, p = 0.588). Seizure types were predominantly generalized with different types of absence seizures in 46/70 (66%), including absence with eyelid myoclonia in 17/70 (24%), and myoclonic seizures in 46/70 (66%). Other frequently observed seizure types included myoclonic–atonic or atonic seizures in 30/70 (43%) and tonic–clonic seizures (28/70, 40%). Less frequently observed were tonic seizures (10/70, 14%), focal seizures (7/70, 10%), epileptic spasms (6/70, 9%), clonic seizures (3/70, 4%), and febrile seizures (2/70, 3%). Males showed a propensity to epileptic spasms (5/14 [36%] males versus 1/56 [2%] females). One reported male did not have seizures but had epileptiform activity on EEG.30
A striking feature was the occurrence of nonconvulsive status epilepticus (NCSE), with prominent myoclonic and absence components, in 13/66 (20%) patients. The most severely affected individuals had episodes of myoclonic status epilepticus lasting several hours that, in some individuals, occurred nightly (Supplementary video).
Environmental photic stimulation was described as a seizure trigger in 7/70 (10%) patients, with eye closure triggering seizures in 5/70 (7%) patients. Clinical photosensitivity was associated with self-induction of seizures (e.g., flickering of hands in front of the eyes) in 4/70 (6%) individuals. Other seizure triggers included stress, high environmental temperature or fever, and perimenstrual period.
Electroencephalography
EEG data (including 21 video EEG data) were available for 63/70 (90%) patients with epilepsy. The predominant findings were generalized spike wave or polyspike wave discharges in 43/63 (68%) patients (Fig. 1). This was accompanied by paroxysmal fast activity (PFA) in 9/63 (14%) patients. In 18/63 (29%) patients, multifocal, or, more rarely focal, epileptiform discharges were observed. In 22/63 (35%) patients, epileptiform activity markedly increased in sleep. Electroencephalographic photosensitivity was documented in 19/63 (30%) patients, consisting of eye closure sensitivity in 15/63 (24%) and/or sensitivity to intermittent photic stimulation (IPS) in 10/63 (16%). Fixation-off sensitivity occurred in 2/63 (3%). Three males had (modified) hypsarrhythmia.13

Interictal epileptiform abnormalities typically consist of generalized polyspike wave (patients 33 and 35) and diffuse rhythmic fast activity (patients 7 and 11) alone or in combination (patient 26). Polyspike wave complexes may be associated with a myoclonic seizure (arrow) and are often accentuated during non-REM sleep (patient 35). Eye closure sensitivity is a common feature (patients 7, 11, 26 and 33; denoted by OO). In one case (patient 11), subsequent trains of diffuse sharply contoured rhythmic activity (10–11 Hz) on eye closure were accompanied by almost continuous eyelid flutter and impaired awareness for more than 30 minutes, consistent with nonconvulsive status epilepticus.
Epilepsy syndromes in new cohort
We were able to classify the epilepsy syndrome in 42/44 new patients based on the available data. The most frequent epilepsy syndromes were eyelid myoclonia with absence (EMA), described by Jeavons in 10 (24%, all female); myoclonic–atonic epilepsy (MAE), described by Doose in 7 (17%, all female); or patients with DEE overlapping these two syndromes in 5 (12%, 2 male, 3 female). We note that two girls classified as EMA had earlier onset (at 9 months and 1 year) than is usually accepted for this syndrome, though earlier onset has been observed in the setting of ID.32 Further, one female had genetic generalized epilepsy with mild ID. Interestingly, West syndrome was diagnosed in one male and another male had epileptic spasms without hypsarrhythmia. Twelve of 42 (29%) patients had a nonsyndromic, myoclonic DEE, while 5/42 (12%) had an unclassified DEE.
Previously, an epilepsy syndrome has been described in only 5/26 published cases. This included MAE in 2 females,17 EMA in one female,20 West syndrome13,28 and Lennox–Gastaut syndrome in one male each.13 Two additional patients had epileptic spasms and hypsarrhythmia but were not classified as West syndrome.13
Seizure outcome
Of the 51 patients on whom information was available regarding treatment, 39/51 (76%) received three or more antiepileptic drugs (AEDs). Ten additional reported patients were drug-resistant, although the number of AEDs trialed was not documented. At last follow-up, 59/70 (84%) patients with seizures had ongoing seizures, while 11 were seizure-free (7/15; 47% males versus 4/55; 7% females, p = 0.001). Our retrospective analysis found that the AEDs reported most often to be effective, in various combinations, were valproate (21 patients), with or without lamotrigine (10 patients), levetiracetam (14 patients), and ethosuximide (15 patients), consistent with their use in generalized epilepsies.
Neurological examination
Neurological examination was normal in 45/85 (53%) patients. Abnormalities included hypotonia (32/85, 38%), poor coordination or ataxia (14/85, 16%), and hypertonia or spasticity (8/85, 9%). Microcephaly (≤2 SD below the mean) was more frequent in males (8/24; 33%) than females (6/60; 10%), although analysis of index patients alone did not show a significant difference (p = 0.120).
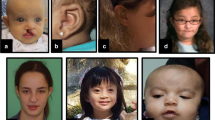
Dysmorphology including automated facial analysis
Information on dysmorphic features, including clinical pictures of 29 patients (11 published, Fig. 2), was available for 85/86 patients. Dysmorphic features were observed in 40/86 (47%) patients and were more frequent in males (16/23, 70%) than females (24/63, 38%, p = 0.032). When reviewing pictures of patients, most had subtle dysmorphic features, with hooded eyelids the most frequent characteristic (15/26; two cases from Kuroda et al.21 omitted due to ethnicity).21 Fewer than 10% of patients had other features such as abnormal ear placement, open hypotonic mouth, and narrow forehead.

Pictures of new patients with NEXMIF encephalopathy, and the first photograph of a reported patient.20 All patients are female, apart from male cases 1, 2, and 14. More frequent features include hooded eyelids, abnormal placement of the ears, open hypotonic mouth, thin upper lip, and narrow forehead.
Automated facial analysis of dysmorphic features could not distinguish our patient cohort from unaffected controls (whole cohort: area under the curve [AUC] of 0.617 [p < 0.132]; male cohort: AUC of 0.607 [p < 0.245]; female cohort: AUC of 0.654 [p < 0.177]) or from our previously reported SYNGAP1 cohort (whole cohort: AUC of 0.643 [p < 0.132]; male cohort: AUC of 0.417 [p < 0.546]; female cohort: AUC of 0.647 [p < 0.207]).33 (See Supplementary methods). Overall, no distinctive facial gestalt was identified clinically or via automated analysis.
Neuroimaging
Brain imaging was available for 78/87 patients. In 68/78 (87%), the MRI (performed at age 7 months to 25 years) was normal or showed minor findings. Ten of 78 (13%) patients had abnormalities, including regional or generalized brain atrophy in 5/78 (6%) (Supplementary Tables S2 and S3). Ten brain MRIs were available for analysis by a pediatric neuroradiologist (S. Mandelstam), and did not reveal any recurrent features (Supplementary Table S2).
Additional comorbidities
Additional comorbidities were identified in 62/86 (72%) patients. These most frequently affected the gastrointestinal system, with infantile gastroesophageal reflux occurring in 19/86 (22%) patients. Pre- or postnatal growth retardation (≤2 SD below the mean) was reported in 13/86 (15%) patients, more often affecting males (9/24, 38%) than females (4/62; 6%) with 11/13 having severe to profound ID. In contrast, about half of the adult patients were overweight or obese (11/18 [61%] females and 2/5 [40%] males). Strabismus was observed in 11/86 (13%; 9 male) patients. Dysfunction of the hypothalamic–pituitary axis occurred in 7/86 (8%; 4 females) patients: 1 had hypogonadotropic hypogonadism, 1 central hypothyroidism, 2 precocious puberty, 1 primary amenorrhea, and 2 delayed bone age.
Molecular genetics
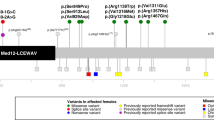
Our cohort of 87 patients (78 families) had 58 unique NEXMIF variants including 28 stop gains, 24 frameshift variants, and 6 larger structural variants (Fig. 3). Thirty variants were novel. Ten variants were recurrent; five were new recurrent variants first identified in our cohort: p.(Trp112*) (n = 2), p.(Met295Valfs*2) (n = 2), p.(Tyr964*) (n = 2), p.(Ser995*) (n = 2), and p.(Asn1153Lysfs*8) (n = 4).

Genomic organization of NEXMIF with location of all pathogenic and likely pathogenic NEXMIF variants (based on Fig. 1 in de Lange et al.17). Coding regions are indicated in blue, untranslated regions in gray. All exons are shown at scale apart from exon 4, which continues as illustrated by the arrow. Previously reported variants are shown above the figure and newly identified variants below. Variants reported only in females are indicated in red, only in males in blue, and variants reported in both males and females in green. Recurrent variants are in bold; the number of independent families carrying the variant is shown in parentheses. Two variants of uncertain significance are indicated in grey.
Fifty-nine of 78 (76%) families had de novo variants (51 females, 8 males) and 10 families had maternally inherited variants (new families with >2 affected individuals in Supplementary Fig. 1). Five of ten families included affected males only (and unaffected heterozygous females), and in five both females and males were affected. Half of the pathogenic small variants seen in families were also seen as de novo variants in sporadic cases. For three patients, only one parent was available and was shown to be negative for their child’s variant. For three patients, parental DNA was not available.
Mosaicism was identified in blood-derived DNA in two male probands. Two sisters shared a recurrent variant and parental gonadal mosaicism was hypothesized, but was not detected in their parents’ blood-derived DNA despite deep sequencing (coverage >200×).29
Genotype–phenotype correlation
Pathogenic NEXMIF variants included stopgain, frameshift, and larger structural variants; all were predicted to decrease protein expression. Smaller molecular variants were located in exon 3, the largest protein-encoding exon in NEXMIF. Missense pathogenic variants were not seen.
We did not expect genotype–phenotype correlations between patients with different variants, as all were predicted to lead to nonsense-mediated messenger RNA (mRNA) decay. The only possible exception was p.(Lys1396Argfs*3) as it is located in the last 10% of the coding region and may escape nonsense-mediated decay; however, the phenotype was consistent with our other cases. The main genotype–phenotype correlation in NEXMIF encephalopathy was related to sex, as detailed above. Interestingly, a reported male with a milder phenotype had an exon 2 duplication that led to a 60% reduction in NEXMIF expression, compared with the absence of expression shown in some males.13
XCI analysis was performed on 32/63 females (21 from new cohort, Supplementary Table 2). Seven females had a skewed XCI pattern. Comparing females with skewed versus females with random XCI, and also comparing sisters with skewed versus random XCI (families 4 and 7), there was no correlation between phenotype and XCI status. Two published females showed 100% skewed XCI and loss of NEXMIF expression and thus resembled males at the molecular level: one had a typical male phenotype (severe ID, ASD, no seizures), while the other’s phenotype was consistent with either sex (moderate ID, no seizures, no autistic features). Interestingly, the two mosaic males (patients 2 and 47), whose genotype resembled an affected female, had moderate ID with worse seizure severity than most hemizygous males.
Variants of uncertain significance
Two families with interesting VUS in NEXMIF were referred to our study (pedigrees in Supplementary Fig. 2; phenotypic data in Supplementary Table S2). One family had an inframe deletion p.(Leu1053del) and the other a missense variant p.(Asp519Val) predicted to be pathogenic by different in silico tools (Supplementary document); both variants were absent in gnomAD.24 The inframe deletion was found in a family with three affected siblings and their unaffected mother, aunt, and grandmother. The variant was not found in their healthy brother. The missense variant was found in a family consisting of two half-brothers with ID, generalized epilepsy in one, and their mildly affected mother. Both families showed phenotypes in keeping with the NEXMIF encephalopathy phenotypic spectrum, suggesting that, with further functional evidence, they may be determined to be pathogenic.
DISCUSSION
In this study, we analyze the phenotypic and genetic features of a large cohort of 87 patients (78 families) with NEXMIF encephalopathy, including 46 new patients with 30 novel NEXMIF variants. All but 117 of 87 patients had cognitive impairment, with severe or profound ID in 43% of cases, with a male preponderance (74% males versus 31% females). ASD or autistic features and behavioral problems were common. Other neuropsychiatric features included stereotypies, and attention and hyperactivity problems.
Developmental delay occurred prior to seizure onset in 41% of patients with epilepsy, and 17% of patients did not have seizures. Regression or stagnation was reported in about a third, often coinciding with seizure onset or exacerbation.
Drug-resistant epilepsy was more frequent in females with NEXMIF encephalopathy. Nevertheless, one of our patients with the most severe seizure phenotype was male (supplementary video) and 7/8 males in our newly identified cohort had seizures, in contrast to “only” 8/15 published cases. Ascertainment bias could have skewed the numbers in the original studies, which had fewer males with seizures, and conversely, in our cohort, as our patients were largely referred by epilepsy centers.
Seizure types were predominantly generalized, most frequently comprising myoclonic, absence of multiple types, atonic or myoclonic–atonic, and tonic–clonic seizures. One in five had a history of NCSE, often recurrent, with florid myoclonic components (Supplementary video). The EEG pattern showed irregular, bilaterally synchronous, symmetric spike wave and polyspike wave activity (Fig. 1). Photosensitivity was observed as either a clinical or electroencephalographic phenomenon in about 30% of patients with epilepsy in response to variation in ambient light or intermittent photic stimulation, a feature typically associated with the genetic generalized epilepsies. Eye closure sensitivity on EEG, seen in 24% of NEXMIF epilepsy patients, is the hallmark of EMA.34 It often occurs in individuals with photic sensitivity and involves a distinct pathophysiological network implicating both the occipital cortex and the supplementary motor area.35
NEXMIF encephalopathy presents a distinctive epilepsy syndrome of a myoclonic DEE comprising an overlap between the syndromes of MAE and EMA (Fig. 4). A similar overlap of these specific epilepsy syndromes is seen in patients with pathogenic variants in CHD2,36 SYNGAP1,33 and SLC6A1.37 This suggests that children with this phenotype should be tested for pathogenic variants in these genes. West syndrome or epileptic spasms, while rare, occurred predominantly in males. Several patients showed features reminiscent of Lennox–Gastaut syndrome, such as slow spike wave and generalized paroxysmal fast activity or tonic seizures.

Conceptual figure showing epilepsy syndromes most frequently associated with NEXMIF encephalopathy and their frequency in females versus males. DEE developmental and epileptic encephalopathies, EMA eyelid myoclonia with absence, MAE myoclonic–atonic epilepsy.
Comorbidities were common in NEXMIF encephalopathy, including infantile hypotonia, ataxia, microcephaly, gastroesophageal reflux disease, strabismus, and dysmorphic features. Detailed clinical and digital analysis did not distinguish a characteristic facies. Growth retardation was more frequently observed in males, whereas obesity was noted in adult females. We observed endocrinological problems, including dysfunction of the hypothalamic–pituitary axis, in 8% of our cohort. As this is a small number, it may be serendipitous and unrelated to NEXMIF encephalopathy.
NEXMIF is an X-linked gene that is thought to play a role in neuronal morphogenesis, migration, and synapse formation, underscoring its critical function in normal brain development.8,9,10,11 NEXMIF encephalopathy is caused by de novo or maternally inherited variants leading to a premature stop codon or larger structural variants. As such, the underlying mechanism is likely loss of function, ranging from total loss of function with no protein expression in hemizygous males, to partial loss of function with a variable decrease in protein expression in females due to XCI. This is supported by the constraint metrics in gnomAD that show that NEXMIF is loss-of-function intolerant (probability of loss of function intolerance [pLI] 1.00, o/e 0.06 [CI 0.03–0.2], http://gnomad.broadinstitute.org/ gene/ENSG00000050030:).24 Further support can be drawn from the recent Nexmif/Kidlia knockout mouse model that recapitulated some of the human disease traits, such as learning deficits, decreased sociability, and seizures.11 The role of inframe deletions and missense variants is unclear, albeit intriguing given the families described above, and requires interrogation with functional studies.
The most obvious genotype–phenotype correlation was that males were more severely affected than females. Males had more severe impairment with three-quarters having severe to profound ID. Epilepsy was more common in females, with females more likely to have EMA and MAE while males accounted for most cases of epileptic spasms. The majority of females had random XCI, not supporting the hypothesis that the XCI pattern in blood explains the phenotypic variation in females. Nevertheless, correlation of XCI status between blood and brain is uncertain, and we are not accessing the best tissue to explain neurological disease.
Other factors such as modifying genes and genetic background likely also influence phenotypic variability. The hypothesis of cellular interference has been invoked in PCHD19 X-linked clustering epilepsy.4 NEXMIF, like PCDH19, is involved in cell–cell adhesion and neuronal migration.8,9,10,17 Thus cellular mosaicism, occurring in females through XCI, could disrupt normal intercellular communication and neuronal networks, potentially predisposing females to epilepsy. This hypothesis is supported by our two males with somatic mosaic NEXMIF encephalopathy, who only had moderate ID and drug-resistant epilepsy, more closely resembling females with NEXMIF encephalopathy.
NEXMIF encephalopathy was initially described as an X-linked disorder in males.12,13 With the identification of additional patients, 72% of cases are now female. A similar skewed sex distribution is observed in other dominant X-linked disorders, such as Rett syndrome associated with MECP2 pathogenic variants. Different hypotheses could explain this skewed sex distribution for an X-linked disease:38 (1) pathogenic variants increase the risk of prenatal lethality in males; (2) pathogenic variants occur predominantly in male germ cells, resulting in affected daughters, as occurs in Rett syndrome,39 or (3) females have a statistically higher chance of a de novo X-linked variant because they have two X-chromosomes.
The majority of patients with NEXMIF encephalopathy had drug-resistant epilepsy. Future efforts to target precision therapies for NEXMIF encephalopathy must also focus on the developmental aspects and comorbidities of this severe disease, in addition to the seizure disorder. In Rett syndrome due to MECP2 pathogenic variants, therapeutic strategies targeting XCI to upregulate gene expression are underway.40 Similar strategies could be considered for females with NEXMIF pathogenic variants. In males, carrying only the mutant NEXMIF allele, different molecular approaches will need to be considered.
NEXMIF encephalopathy typically presents with one of two pictures: most commonly, a myoclonic developmental and epileptic encephalopathy associated with drug-resistant generalized epilepsy and less frequently, a developmental encephalopathy without epilepsy, more frequent in males. The NEXMIF epilepsy syndrome is an overlap of MAE and EMA. This X-linked loss-of-function disorder presents with a broad phenotypic spectrum in females, in contrast to a more severe picture in males.
References
Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–521.
Howell KB, Eggers S, Dalziel K, et al. A population-based cost-effectiveness study of early genetic testing in severe epilepsies of infancy. Epilepsia. 2018;59:1177–1187.
Heyne HO, Singh T, Stamberger H, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet. 2018;50:1048–1053.
Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. 2008;40:776–781.
Mignot C, McMahon AC, Bar C, et al. IQSEC2-related encephalopathy in males and females: a comparative study including 37 novel patients. Genet Med. 2019;21:837–849.
Migeon BR. The role of X inactivation and cellular mosaicism in women’s health and sex-specific diseases. JAMA. 2006;295:1428–1433.
Plenge RM, Stevenson RA, Lubs HA, Schwartz CE, Willard HF. Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders. Am J Hum Genet. 2002;71:168–173.
Gilbert J, Man HY. The X-linked autism protein KIAA2022/KIDLIA regulates neurite outgrowth via N-cadherin and delta-catenin signaling. eNeuro. 2016;3:ENEURO.0238-16.2016.
Magome T, Hattori T, Taniguchi M, et al. XLMR protein related to neurite extension (Xpn/KIAA2022) regulates cell–cell and cell-matrix adhesion and migration. Neurochem Int. 2013;63:561–569.
Ishikawa T, Miyata S, Koyama Y, et al. Transient expression of Xpn, an XLMR protein related to neurite extension, during brain development and participation in neurite outgrowth. Neuroscience. 2012;214:181–191.
Gilbert J, O’Connor M, Templet S, et al. NEXMIF/KIDLIA knock-out mouse demonstrates autism-like behaviors, memory deficits, and impairments in synapse formation and function. J Neurosci. 2020;40:237–254.
Cantagrel V, Lossi AM, Boulanger S, et al. Disruption of a new X linked gene highly expressed in brain in a family with two mentally retarded males. J Med Genet. 2004;41:736–742.
Van Maldergem L, Hou Q, Kalscheuer VM, et al. Loss of function of KIAA2022 causes mild to severe intellectual disability with an autism spectrum disorder and impairs neurite outgrowth. Hum Mol Genet. 2013;22:3306–3314.
Athanasakis E, Licastro D, Faletra F, et al. Next generation sequencing in nonsyndromic intellectual disability: from a negative molecular karyotype to a possible causative mutation detection. Am J Med Genet A. 2014;164A:170–176.
Moyses-Oliveira M, Guilherme RS, Meloni VA, et al. X-linked intellectual disability related genes disrupted by balanced X-autosome translocations. Am J Med Genet B Neuropsychiatr Genet. 2015;168:669–677.
Farach LS, Northrup H. KIAA2022 nonsense mutation in a symptomatic female. Am J Med Genet A. 2016;170:703–706.
de Lange IM, Helbig KL, Weckhuysen S, et al. De novo mutations of KIAA2022 in females cause intellectual disability and intractable epilepsy. J Med Genet. 2016;53:850–858.
Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:522–530.
Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-9. Epilepsia. 2010;51:676–685.
Samanta D, Willis E. KIAA2022-related disorders can cause Jeavons (eyelid myoclonia with absence) syndrome. Acta Neurol Belg. 2020;120:205–207.
Kuroda Y, Ohashi I, Naruto T, et al. Delineation of the KIAA2022 mutation phenotype: two patients with X-linked intellectual disability and distinctive features. Am J Med Genet A. 2015;167:1349–1353.
Webster R, Cho MT, Retterer K, et al. De novo loss of function mutations in KIAA2022 are associated with epilepsy and neurodevelopmental delay in females. Clin Genet. 2017;91:756–763.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Charzewska A, Rzonca S, Janeczko M. et al. A duplication of the whole KIAA2022 gene validates the gene role in the pathogenesis of intellectual disability and autism. Clin Genet. 2015;88:297–299.
Borlot F, Regan BM, Bassett AS, Stavropoulos DJ, Andrade DM. Prevalence of pathogenic copy number variation in adults with pediatric-onset epilepsy and intellectual disability. JAMA Neurol. 2017;74:1301–1311.
Lorenzo M, Stolte-Dijkstra I, van Rheenen P, Smith RG, Scheers T, Walia JS. Clinical spectrum of KIAA2022 pathogenic variants in males: case report of two boys with KIAA2022 pathogenic variants and review of the literature. Am J Med Genet A. 2018;176:1455–1462.
Lambert N, Dauve C, Ranza E, et al. Novel NEXMIF pathogenic variant in a boy with severe autistic features, intellectual disability, and epilepsy, and his mildly affected mother. J Hum Genet. 2018;63:847–850.
Myers CT, Hollingsworth G, Muir AM, et al. Parental mosaicism in “de novo” epileptic encephalopathies. N Engl J Med. 2018;378:1646–1648.
Alarcon-Martinez T, Khan A, Myers KA. Torpedo maculopathy associated with NEXMIF mutation. Mol Syndromol. 2019;10:229–233.
Gurovich Y, Hanani Y, Bar O, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019;25:60–64.
Caraballo RH, Fontana E, Darra F, et al. A study of 63 cases with eyelid myoclonia with or without absences: type of seizure or an epileptic syndrome? Seizure. 2009;18:440–445.
Vlaskamp DRM, Shaw BJ, Burgess R, et al. SYNGAP1 encephalopathy: a distinctive generalized developmental and epileptic encephalopathy. Neurology. 2019;92:e96–e107.
Rubboli G, Gardella E, Capovilla G. Idiopathic generalized epilepsy (IGE) syndromes in development: IGE with absences of early childhood, IGE with phantom absences, and perioral myoclonia with absences. Epilepsia. 2009;50 Suppl 5:24–28.
Vaudano AE, Ruggieri A, Tondelli M, et al. The visual system in eyelid myoclonia with absences. Ann Neurol. 2014;76:412–427.
Thomas RH, Zhang LM, Carvill GL, et al. CHD2 myoclonic encephalopathy is frequently associated with self-induced seizures. Neurology. 2015;84:951–958.
Johannesen KM, Gardella E, Linnankivi T, et al. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia. 2018;59:389–402.
Thomas GH. High male:female ratio of germ-line mutations: an alternative explanation for postulated gestational lethality in males in X-linked dominant disorders. Am J Hum Genet. 1996;58:1364–1368.
Girard M, Couvert P, Carrie A, et al. Parental origin of de novo MECP2 mutations in Rett syndrome. Eur J Hum Genet. 2001;9:231–236.
Carrette LLG, Wang CY, Wei C, et al. A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc Natl Acad Sci U S A. 2018;115:E668–E675.
Acknowledgements
We thank the patients and their families for participating in our research. We further want to acknowledge the Epi25 Consortium for providing a platform for exome sequencing for epilepsy patients that led to a diagnosis for some of the patients included in this study. H.S. was PhD fellow of the Fund for Scientific Research Flanders (1125416N). H.C.M. received support from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) (R01NS069605). D.M.A. received funding from EpLink, of the Ontario Brain Institute for array comparative genomic hybridization (aCGH) analysis. This study was supported by the Key Research Project of the Ministry of Science and Technology of China (2016YFC0904400 and 2016YFC0904401; through J.Z. and Y.A.Z.), Cure Kids New Zealand (through L.G.S.) and the Victorian Government’s Operational Infrastructure Support Program and the Australian Government National Health and Medical Research Council (NHMRC) Independent Medical Research Institutes Infrastructure Support Scheme (IRIISS) (through M.B.). The work was generated within ITHACA (Intellectual disability, TeleHealth, And Congenital Anomalies), European Reference Network on Rare Congenital Malformations, and Rare Intellectual Disability.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
R.G. has acted as an investigator for studies with Zogenix, Biocodex, Biomarin, UCB, Angelini, and Eisai Inc. He has been a speaker and on advisory boards for Zogenix, Biocodex, Novartis, Biomarin, GW Pharma, and Biocodex; serves/has served on the editorial boards of Epilepsia, Progress in Epileptic Disorders, Neuropediatrics, Journal of Child Neurology, Seizure, BMC Medical Genetics, Topics in Epilepsy, and Neurology. He receives/has received research support from the Italian Ministry of Health, The European Community, The Tuscany Region, the Mariani Foundation, The Pisa Foundation, The Fund of Epilepsy, The GKT Special Trustees, The Italian Federation for Epilepsy, and The Italian Association for Epilepsy. I.E.S. serves/has served on the editorial boards of the Annals of Neurology, Neurology, and Epileptic Disorders; may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; has a patent for SCN1A testing held by Bionomics, Inc., and licensed to various diagnostic companies; has a patent molecular diagnostic/theranostic target for benign familial infantile epilepsy (BFIE) [PRRT2] 2011904493 & 2012900190 and PCT/AU2012/001321 (TECH ID:2012-009) with royalties paid. She has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, and Xenon Pharmaceuticals; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, Biomarin, and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx, GW Pharma, UCB, Eisai, Anavex Life Scienand Marinus; and has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics, and UCB. She receives/has received research support from the National Health and Medical Research Council of Australia, Health Research Council of New Zealand, CURE, Australian Epilepsy Research Fund, March of Dimes and NIH/NINDS. D.M.A. has consulted for Eisai, UCB, and is part of the MAB of Stoke Therapeutics and DSF. B.G. has received consultancy fees from GW Pharmaceuticals companies, Zogenix, and Ovid/Takeda; and is a principal investigator for GW Research Ltd, Zogenix, LivaNova, Marinus Pharmaceuticals, and UCB. L.G.S. reports grants from Health Research Council of New Zealand, grants from Cure Kids New Zealand, during the conduct of the study; grants and personal fees from Zynerba, other from Sequirus, other from Nutricia, outside the submitted work. S.M.Z. has received research funding from Epilepsy Research UK, UCB Pharma, Glasgow Children’s Hospital Charity, Dravet Syndrome UK. He serves as Editor-in-Chief of European Journal of Pediatric Neurology for which he receives an honorarium from Elsevier Ltd. He has participated in educational symposia/advisory boards/consulted for GW Pharma, Zogenix Ltd, Biocodex, Nutricia, and Encoded Genomics. The other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Stamberger, H., Hammer, T.B., Gardella, E. et al. NEXMIF encephalopathy: an X-linked disorder with male and female phenotypic patterns. Genet Med 23, 363–373 (2021). https://doi.org/10.1038/s41436-020-00988-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-00988-9
Keywords
This article is cited by
-
Epilepsy and Other Phenotypic Features of X-Linked Intellectual Disability Due to Mutations in the KIAA2022 Gene
Neuroscience and Behavioral Physiology (2023)
-
Loss of Nexmif results in the expression of phenotypic variability and loss of genomic integrity
Scientific Reports (2022)
-
Systematic analysis and prediction of genes associated with monogenic disorders on human chromosome X
Nature Communications (2022)
