
Abstract
Purpose
We report prospective clinical investigations of children affected with periodontal Ehlers–Danlos syndrome (pEDS). The main clinical features of pEDS in adults are early severe periodontitis, generalized lack of attached gingiva, and pretibial hemosiderin plaques due to dominant pathogenic variants in the C1R or C1S genes.
Methods
Nineteen children with a parent diagnosed with molecularly confirmed pEDS underwent physical examination including oral and radiological investigations followed by genetic testing.
Results
The only consistent manifestation of pEDS in childhood was a characteristic gingival phenotype: generalized lack of attached gingiva. All children with this gingival phenotype had inherited the familial pathogenic variant (n = 12) whereas the gingival phenotype was absent in children without the familial pathogenic variant (n = 7). Easy bruising was reported in eight affected and zero unaffected children. Other manifestations of pEDS were rarely present in children. Only 2/12 affected children aged 8 and 13 years fulfilled the clinical criteria for pEDS.
Conclusion
Generalized lack of attached gingiva is a pathognomonic feature of pEDS and the only clinical finding that is consistently present in affected adults and children. This is important because an early diagnosis may facilitate better dental hygiene in childhood, which may be essential to prevent early dental loss.
Similar content being viewed by others

INTRODUCTION
Ehlers–Danlos syndromes (EDS) are a group of clinically and genetically heterogeneous connective tissue disorders.1,2,3 Their overlapping features include joint hypermobility, skin and vascular fragility, and generalized connective tissue friability.4 Periodontal Ehlers–Danlos syndrome (pEDS) is characterized by distinct oral manifestations consisting of early severe periodontal bone loss leading to gingival recession and premature loss of teeth. Heterozygous pathogenic variants in C1R (MIM 613785) or C1S (MIM 120580) encoding complement 1 subcomponents r and s, respectively (C1R; C1S) have been identified to cause pEDS. Activation of C1R and C1S is the first step of the classical complement cascade, a major antimicrobial pathway of the innate immune system. Individuals with pEDS have heterozygous missense or in-frame insertion/deletion variants in C1R or C1S. The pathogenesis of pEDS is only partly understood; experimental evidence suggests that it is linked to secretion or release of active C1r serine protease in the extracellular space.5,6 This mechanism may cause gingival hyperinflammation in response to mild biofilm accumulation, and subsequently rapidly progressing periodontal destruction. A broad spectrum of C1R and C1S substrate specificities could explain the various noninflammatory clinical symptoms in pEDS.6
In the 2017 classification of Ehlers–Danlos syndromes, clinical criteria suggestive for periodontal EDS (pEDS) were defined.2 Major criteria are (1) severe and intractable periodontitis of early onset in childhood or adolescence, (2) lack of attached gingiva, (3) pretibial plaques, and (4) family history of a first-degree relative who meets clinical criteria.2 Three major criteria and one of the minor criteria must be fulfilled. Minor criteria consist of easy bruising, joint hypermobility (mostly distal joints), skin hyperextensibility and fragility, abnormal scarring, increased rate of infections, hernias, marfanoid facial features, acrogeria, and prominent vasculature.2 A clinical diagnosis of pEDS should be confirmed with genetic testing.
Periodontitis is characterized by progressive destruction of the tooth-supporting tissues, namely alveolar bone, periodontal ligament, root cementum, and gingival attachment (see Fig. 1). The diagnosis is based on alveolar bone loss visible on radiographs, clinical measurement of attachment loss, and gingival bleeding. Progressing alveolar bone loss finally leads to dental loss. The 2017 classification of periodontal and peri-implant diseases and conditions distinguishes between periodontitis as a common and chronic multifactorial inflammatory disease associated with dysbiotic plaque biofilms, and periodontitis as a manifestation of systemic diseases.7 The latter includes the diagnosis of pEDS and other mainly rare systemic conditions associated with severe periodontal destruction.8 Complete tooth loss in pEDS has been reported to occur at a mean age of 20 years.9 The youngest individual with complete dental loss was 14 years old; however, some individuals older than 60 years were noted to have kept at least several teeth. Potentially, this may be due to intensive periodontal treatment or other influencing factors that are currently unknown.

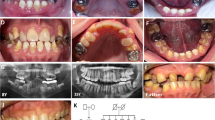
(a) The tooth is composed of enamel, dentine, cementum, and the pulp. Enamel is the outer layer of the tooth crown, an epithelially derived and highly (approximately 96%) mineralized tissue with traces of noncollagenous proteins (mainly amelogenins).17 Dentine forms the bulk of the tooth. It is highly mineralized and composed of approximately 70% hydroxyapatite crystals embedded in a three-dimensional collagenous network (consisting of mainly type I collagen with traces of type III and V collagen) associated with noncollagenous proteins and proteoglycans. The dental pulp is a highly vascularized and innervated connective tissue characterized by its specific anatomical location;18 types I, III, and V collagen represent 56%, 41%, and 2% of the total collagen, respectively.19 The periodontal ligament belongs to the tooth-supporting tissues and anchors the tooth root to the alveolar bone. It is a highly specialized connective tissue and contains well-defined collagen fiber bundles embedded in ground substance (collagen types I, III, and XII).17 The cementum covers the root and anchors the periodontal ligament fibers to the tooth. Its main function is tooth attachment. It consists of approximately 50% inorganic hydroxyapatite. Collagen type I with small amounts of collagen types III and XII and noncollageneous proteins including several proteoglycans form the organic matrix. The attached gingiva is keratinized and tightly bound to the periosteum through collagen type I fibers. (b) Periodontitis is a chronic multifactorial inflammatory disease associated with dysbiotic plaque biofilms and characterized by progressive destruction of the tooth-supporting tissues,7 alveolar bone, periodontal ligament, root cementum, and gingival attachement. As a consequence, periodontal pockets (the gap between the tooth and the gums; marked with a white arrow) develop, extending abnormally deep apically to the original level of the resorbed alveolar crest. Progressing alveolar bone loss finally leads to dental loss. The community periodontal index of treatment needs (CPITN) is a periodontal screening tool measuring periodontal pocket depths. CPITN grade 0 resembles healthy periodontal conditions. CPITN grades 1 and 2 resemble plaque-induced gingivitis without periodontal destruction and indicating the need of oral hygiene instructions and in grade 2 additional removal of plaque retentive factors. CPITN grade 3 and 4 resemble mild and severe periodontitis and indicating more detailed examination of periodontal condition and periodontal treatment.13
Lack of attached gingiva (for description see Fig. 2a) and oral tissue fragility have been reported in 16 affected adult individuals and 3 affected children.10,11,12 For the remaining 120 individuals described to date no data or intraoral photographs are available.

(a) The gingiva is subdivided into the nonattached free gingiva (which includes the gingival margin and the papillae) and the attached gingiva. The attached gingiva is tightly and unmovably bound to the periosteum via collagen type I fibers. The gingiva is keratinized and thicker than the mucosa to provide mechanical protection. The border between attached gingiva and mucosa constitutes the mucogingival junction. In vivo, it is sometimes visible as a white line (see d–g lower jaw). The oral mucosa is loosely connected to the periosteum and nonkeratinized. It is movable, thin, more fragile, translucent, and blood vessels are visible. (b–g) In all unaffected healthy siblings the attached gingiva was present. This is most clearly visible in the upper jaw.
Up to now, clinical manifestations have been studied in adult patient cohorts only, and symptoms in childhood were retrospectively collated.9 In the current study we report prospective clinical investigations of a cohort of children with a 50% a priori risk of having inherited pEDS. We aimed to define clinical features of pEDS in childhood to determine clinical criteria to distinguish affected from unaffected children. An early diagnosis of pEDS is important to ensure correct dental management that might prevent early loss of teeth.
MATERIALS AND METHODS
Ethics statement
The study was conducted with the probands’ understanding and full written consent as part of the Biobank for Rare Diseases approved by the Ethics Committee of the Medical University Innsbruck, Austria (studies UN4501 and 1074/2017), and in accordance with the Declaration of Helsinki of 1975 as revised in 2013. Physical examination and genetic analyses also served clinical diagnostic purposes for guiding management of affected children. For publication of patient photos, consent to publish was obtained from the legal representatives of the children.
Clinical and genetic investigations
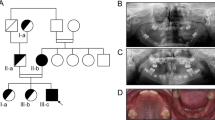
Nineteen children from six families (A–F), each with a 50% chance of having inherited pEDS from a parent with a molecularly confirmed diagnosis of pEDS, were included. All children underwent physical examination comprising oral investigations and subsequent genetic testing for the familial variant. The clinical investigation list and the questionnaire for reporting on pEDS have previously been published (Kapferer-Seebacher et al.,9 Supplement 1 “Investigating periodontal EDS” and Supplement 3 “Questionnaire periodontal EDS”). In summary, clinical investigations included complete physical examination with assessment of joint hypermobility and skin features and clinical photographs. Oral investigations included the community periodontal index of treatment needs (CPITN; explanation see Fig. 1),13 orthopantomogram, and intraoral photographs. The diagnosis of periodontitis was based on radiologic evidence of alveolar bone loss (≥10%) and/or CPITN grade 3 or 4. The questionnaire included questions regarding previous dental treatment and risk factors for periodontal disease; joint and skin features; other features previously reported in people with pEDS such as recurrent infections, hoarse voice, organ ruptures; systemic conditions previously reported with pEDS or possibly correlated with the classical complement pathway. Family A was investigated at the Medical University of Innsbruck (Austria); families B, C, D were investigated at the National EDS Service, London (United Kingdom); family E was investigated at the VU University Medical Center, Amsterdam (The Netherlands); and family F at the National EDS Service, Sheffield (United Kingdom). All oral investigations were performed by the same periodontal specialist (I.K.-S.).
Genomic DNA was isolated from peripheral blood using standard procedures. Causative variants in C1R (NM_001733.5) had been previously identified in all families; children were tested for the familial variant by polymerase chain reaction (PCR) amplification and Sanger sequencing of the respective C1R exon, using standard methods.
RESULTS
Subjects, families, and genetic results
Twelve children from six families with molecularly confirmed pEDS aged 4 to 13 years showed clinical manifestations suggestive of pEDS and were subsequently confirmed to have inherited the familial pathogenic C1R variant. Seven siblings aged seven months to ten years were clinically and genetically unaffected. Clinical features of each individual can be found in Table 1 and have been described in detail in the Supplementary appendix. The identified familial variants were all identified in the C1R gene as follows: family A: c.149_150delinsAT, p.oVal50Asp; family B: c.689T>C, p.Leu230Pro (not previously reported); family C: c.702_711delinsT, p.Lys235_Leu237del (not previously reported); family D: c.869A>G, p.Asp290Gly; family E: c.926G>T, p.Cys309Phe; family F: c.1113C>G, p.Cys371Trp (not previously reported). The identified C1R variants have been submitted to ClinVar.
The most specific and consistent clinical manifestation of pEDS in childhood was generalized lack of attached gingiva, which allowed for a clinical diagnosis of pEDS (Fig. 3). Lack of attached gingiva was obvious in 11/12 affected children. In one child with excellent oral hygiene the gingival phenotype was difficult to diagnose and could only be verified by rolling the mucosa with a probe gently to the gingival margin (patient A1, Fig. 3i). None of the unaffected children presented with this gingival phenotype, revealing a specificity of 100% for this diagnostic criterion (Fig. 2). Periodontal bone loss diagnosed radiologically and clinically was only evident in the oldest investigated child aged 13 years (F2). Parents of three children reported preliminary loss of primary teeth at age 3 to 4 years (C1, C2, D2); they reported that the “teeth came out with the whole root” which seems to be attributable to periodontitis of primary teeth. Six children (A1, C2, C2, D2, E1, F1) presented with gingival recession (Fig. 3). Gingival inflammation and bleeding was a frequent finding (Fig. 3). We did not observe any other oral abnormalities.

In all affected children the attached gingiva is missing, and the thin and fragile mucosa extends to the free gingival margin and the interdental papillae. The free gingival margin and the papillae are normally keratinized and “thick.” In most individuals the generalized lack of attached gingiva is very clear on the photographs due to the extraordinary thin and translucent mucosa with increased vascular visibility (a, c, d, e, h, j, k); in one child A1 (i) it could only be diagnosed clinically by rolling the mucosa with a probe gently to the gingival margin. Gingival recession with exposed dental roots (= receding gums) was present in children C2 and F1 (a, b). Gingival recession, likely attributable to physiological dentition change, was seen near primary teeth of individuals D2, C2, E1, A1 (d, f, h, i). Gingival inflammation recognizable as red and edematous tissues due to insufficient oral hygiene and a hyperinflammatory reaction typical for pEDS was a frequent finding (b, c, d, e, f, h, j, l).
Parents reported easy bruising in response to mild trauma in 8 of 12 affected children and in none of the unaffected children. Pretibial plaques and joint hypermobility were rare in our group of children with pEDS. Pretibial plaques were present in two affected children aged 8 and 13 years (E1, F2; see Fig. 4). Generalized joint hypermobility with a Beighton score of 6/9 was diagnosed in one affected child (F2). Joint hypermobility of the distal joints and mild generalized hypermobility was not specific (see Table 1) and was also observed in unaffected children. Joint subluxation, joint pain, fragile skin, frequent nose bleeds, eardrum rupture, and recurrent herpes zoster were reported in single affected individuals.

(a) Persistent pretibial plaques were only diagnosed in the oldest individual F2 aged 13 years. (b) One boy (E1) had severe pretibial hematomas that seemed excessive for the reported trauma.
DISCUSSION
Nineteen children from six families with a parent who has a molecularly confirmed diagnosis of pEDS were prospectively assessed through medical history taking and clinical examination including oral (clinical and radiologic) investigations, followed by genetic testing.
According to the 2017 minimal criteria suggestive for a diagnosis of pEDS2 only two children of our present cohort would have been diagnosed with pEDS. This included the 13-year-old individual F2 who already had some periodontal bone loss, lack of attached gingiva, pretibial plaques, a Beighton score of 6/9, and easy bruising, and the 8-year-old individual E1 with lack of attached gingiva, severe pretibial hematomas, and easy bruising. None of the ten other affected children met the criteria for a clinical diagnosis of pEDS. The observation that clinical criteria developed for affected adults are not suitable for the diagnosis in children is shared with many other genetic syndromes. However, a feature of pEDS present in all investigated affected children but not in investigated unaffected children, are strikingly thin and sensitive oral soft tissues with generalized lack of attached gingiva (Figs. 2 and 3). In most individuals this specific phenotype was easily recognized due to the translucent mucosa with increased vascular visibility. In one child (A1) it could only be diagnosed by rolling the moveable mucosa with a probe gently to the gingival margin. This suggests that diagnosing this specific feature in rare cases may be challenging even for dentists if they have not had special training in periodontology. In our small group of children the periodontal assessment for generalized lack of attached gingiva had a specificity of 100% and was not age related.
In the general population the width of attached gingiva varies among oral regions and individuals, and increases with age.14 It is significantly higher in the permanent (3.4 ± 0.36 mm) than in the primary dentition (2.17 ± 0.71 mm), and significantly higher in the maxilla than in the mandibula.14,15 Localized lack of attached gingiva is a common finding, e.g., in the mandibular premolars or lower front teeth. However, generalized lack of attached gingiva seems to be pathognomonic for pEDS as, to the best of our knowledge, it has not been reported in any other condition up to now.
Gingival recession is a common finding in adults with pEDS and it is attributed to both inflammatory destruction of the tooth-supporting tissues and thin gums that recede in response to mechanical trauma. In the mixed dentition diagnosing gingival recession is prone to error. This is because permanent teeth have longer crowns than the primary teeth and as such it may seem that there is gingival recession. However, this not the case as the gingival margin lies in the correct position on the cementoenamel junction. This applies to individuals D, E, F, G, H, and I (Fig. 3).
In most affected children in our cohort other clinical manifestations of pEDS were mild or not yet present. Pretibial plaques, a major criterion in adults, does not appear suitable for the diagnosis in childhood as persistent hemosiderinic discolorations were only diagnosed in the oldest individual (F2, aged 13 years). One boy (E1, Fig. 4) of our cohort showed recent severe pretibial hematomas that seemed excessive for the reported trauma. The minor criteria easy bruising and gum bleeding were difficult to evaluate as they are nonspecific and subjective. Gum bleeding, a sign of gingival inflammation, is very frequent in the general population due to insufficient plaque control. Ease of bruising is also subjective but bruising in atypical anatomical regions (like the cheeks in individual E1) in combination with other features may point to an underlying genetic condition such as pEDS. Except for the oldest child (F2), none of the children with pEDS in our cohort showed generalized joint hypermobility, and 6/11 affected children had a Beighton score of 0/9.
In the literature, different forms of periodontitis have been distinguished. “Chronic” or “adult” periodontitis represent the forms of slowly progressive disease, and “aggressive” periodontitis represents a diverse group of highly destructive forms of periodontitis affecting primarily young individuals and including conditions formerly known as “early-onset,” “rapidly progressing,” “prepubertal,” and “juvenile” periodontitis.16 The current classification of periodontal and peri-implant diseases no longer distinguishes between these categories due to substantial overlap, lack of clear pathobiology-based distinction, diagnostic imprecision, and implementation difficulties.7 It is recommended that periodontitis as a direct manifestation of a systemic disease—such as pEDS—should follow the classification of the primary disease.7 To distinguish periodontal EDS from generalized periodontitis affecting young individuals the diagnostic criteria of the 2017 classification of Ehlers–Danlos syndromes should be applied and recognition of complete lack of attached gingiva as a pathognomonic features in children and adults with pEDS should be recognized. Genetic testing in children is recommended when there is a family history of a first-degree relative who meets clinical criteria for pEDS and/or the clinical feature lack of attached gingiva as we have shown that this is pathognomonic of pEDS. The other major clinical features of pEDS are pretibial plaques and periodontitis of early onset, which were observed to be often not present in young children.
Premature loss of primary teeth may be an early sign of pEDS and was reported in three children of our cohort. The average age of onset of periodontal bone loss is still unknown but most probably is in the teens and depends on dental hygiene. Based on retrospectively collected data (medical history) we previously reported a median age of 14 years (range of 2–35 years) for the first diagnosis of periodontal bone loss in pEDS.12 Clinical and radiological investigations in teenagers would be required to determine more precisely the age of onset of rapidly progressing periodontal destruction.
Current dental treatment of pEDS mainly consists of strict biofilm management to stop the plaque-associated periodontal hyperinflammation and probably the subsequent bone and tooth loss. Even though periodontal bone loss is not evident in young children, it is anticipated that an early diagnosis of pEDS would enable an early start of a dental prophylaxis with—in the authors’ opinion—a likely improved chance to prevent or reduce dental loss. Patients will need recurrent training in tooth-brushing and interdental cleaning measures to achieve excellent oral hygiene. For this purpose, we have developed information leaflets with recommendations for parents and dentists to maintain oral hygiene in children with pEDS (Supplementary appendix).
Conclusion
The diagnosis of pEDS can be made in early childhood, based on strikingly thin and sensitive oral soft tissues with a generalized lack of attached gingiva. Other diagnostic criteria of pEDS established in adults are not suitable for children. Early diagnosis is essential for initiating appropriate oral hygiene at an early age.
References
Morlino S, Micale L, Ritelli M, et al. COL1-related overlap disorder: a novel connective tissue disorder incorporating the osteogenesis imperfecta/Ehlers–Danlos syndrome overlap. Clin Genet. 2020;97:396–406.
Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8–26.
Blackburn PR, Xu Z, Tumelty KE, et al. Bi-allelic alterations in AEBP1 lead to defective collagen assembly and connective tissue structure resulting in a variant of Ehlers–Danlos syndrome. Am J Hum Genet. 2018;102:696–705.
Malfait F. Vascular aspects of the Ehlers–Danlos syndromes. Matrix Biol. 2018;71-72:380–395.
Grobner R, Kapferer-Seebacher I, Amberger A, et al. C1R mutations trigger constitutive complement 1 activation in periodontal Ehlers–Danlos syndrome. Front Immunol. 2019;10:2537.
Bally I, Dalonneau F, Chouquet A, et al. Two different missense C1S mutations, associated to periodontal Ehlers–Danlos syndrome, lead to identical molecular outcomes. Front Immunol. 2019;10:2962.
Papapanou PN, Sanz M, Buduneli N, et al. Periodontitis: consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Periodontol. 2018;89 Suppl 1:S173–S182.
Jepsen S, Caton JG, Albandar JM, et al. Periodontal manifestations of systemic diseases and developmental and acquired conditions: consensus report of workgroup 3 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Clin Periodontol. 2018;45 Suppl 20:S219–S229.
Kapferer-Seebacher I, Lundberg P, Malfait F, Zschocke J. Periodontal manifestations of Ehlers–Danlos syndromes: a systematic review. J Clin Periodontol. 2017;44:1088–1100.
Mataix J, Banuls J, Munoz C, Bermejo A, Climent JM. Periodontal Ehlers–Danlos syndrome associated with type III and I collagen deficiencies. Br J Dermatol. 2008;158:825–830.
Cunniff C, Williamson-Kruse L. Ehlers–Danlos syndrome, type VIII presenting with periodontitis and prolonged bleeding time. Clin Dysmorphol. 1995;4:145–149.
Kapferer-Seebacher I, Pepin M, Werner R, et al. Periodontal Ehlers–Danlos syndrome is caused by mutations in C1R and C1S, which encode subcomponents C1r and C1s of complement. Am J Hum Genet. 2016;99:1005–1014.
Ainamo J, Barmes D, Beagrie G, Cutress T, Martin J, Sardo-Infirri J. Development of the World Health Organization (WHO) community periodontal index of treatment needs (CPITN). Int Dent J. 1982;32:281–291.
Ainamo J, Talari A. The increase with age of the width of attached gingiva. J Periodontal Res. 1976;11:182–188.
Vandana KL, Shivani S, Savitha B, Vivek HP. Assessment of gingival sulcus depth, width of attached gingiva, and gingival thickness in primary, mixed, and permanent dentition. J Dent Res Rev. 2017;4:8.
Armitage GC. Development of a classification system for periodontal diseases and conditions. Ann Periodontol. 1999;4:1–6.
Nanci A. Structure of the oral tissues. In: Ten Cate´s Histology. 7th ed. St. Louis: Mosby Elsevier; 2008. p. 3.
Linde A. The extracellular matrix of the dental pulp and dentin. J Dent Res. 1985;64:523–529.
Tsuzaki M, Yamauchi M, Mechanic GL. Bovine dental pulp collagens: characterization of types III and V collagen. Arch Oral Biol. 1990;35:195–200.
Acknowledgements
We thank the families with periodontal EDS for their kind cooperation. We grateful to J. Leary for recontacting families with a (suspected) diagnosis of pEDS in the South of the UK, and to A. Rinner for providing X-rays and photographs of some Austrian patients.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Kapferer-Seebacher, I., Oakley-Hannibal, E., Lepperdinger, U. et al. Prospective clinical investigations of children with periodontal Ehlers–Danlos syndrome identify generalized lack of attached gingiva as a pathognomonic feature. Genet Med 23, 316–322 (2021). https://doi.org/10.1038/s41436-020-00985-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-00985-y
Keywords
This article is cited by
-
An exemplary model of genetic counselling for highly specialised services
Journal of Community Genetics (2023)
