
Abstract
Purpose
Biallelic variants in TBC1D24, which encodes a protein that regulates vesicular transport, are frequently identified in patients with DOORS (deafness, onychodystrophy, osteodystrophy, intellectual disability [previously referred to as mental retardation], and seizures) syndrome. The aim of the study was to identify a genetic cause in families with DOORS syndrome and without a TBC1D24 variant.
Methods
Exome or Sanger sequencing was performed in individuals with a clinical diagnosis of DOORS syndrome without TBC1D24 variants.
Results
We identified the same truncating variant in ATP6V1B2 (NM_001693.4:c.1516C>T; p.Arg506*) in nine individuals from eight unrelated families with DOORS syndrome. This variant was already reported in individuals with dominant deafness onychodystrophy (DDOD) syndrome. Deafness was present in all individuals, along with onychodystrophy and abnormal fingers and/or toes. All families but one had developmental delay or intellectual disability and five individuals had epilepsy. We also describe two additional families with DDOD syndrome in whom the same variant was found.
Conclusion
We expand the phenotype associated with ATP6V1B2 and propose another causal gene for DOORS syndrome. This finding suggests that DDOD and DOORS syndromes might lie on a spectrum of clinically and molecularly related conditions.
Similar content being viewed by others


INTRODUCTION
Biallelic variants in TBC1D24 (MIM 613577), which encodes a protein that regulates vesicular transport, have been identified in half of the patients with DOORS syndrome (MIM 220500); however, no causal gene has been identified in the remaining half.1 DOORS syndrome stands for deafness, onychodystrophy, osteodystrophy, developmental delay and/or intellectual disability—previously referred as mental retardation—and seizures. Dominant deafness onychodystrophy (DDOD; MIM 124480) is a condition that partially overlaps with DOORS syndrome, without intellectual disability and seizures. The mode of inheritance—autosomal recessive in DOORS syndrome and autosomal dominant in DDOD syndrome—also differs. DDOD syndrome is caused by a truncating variant in ATP6V1B2 (MIM 606939), encoding a subunit of the vacuolar (V)-ATPase responsible for the acidification of intracellular organelles.2,3 We had previously reported a different variant in ATP6V1B2 causing Zimmermann–Laband syndrome (MIM 616455), characterized by gingival hyperplasia, intellectual disability, and nail and phalangeal abnormalities.4 Two other ATP6V1B2 variants were reported: one in a patient with intellectual disability, hypotonia, microcephaly, and seizures,5 and another with epilepsy and mild gingival and nail anomalies. However, their phenotypes were otherwise not consistent with DOORS syndrome.6
Here, we report nine individuals from eight unrelated families with a clinical diagnosis of DOORS syndrome and no variant in TBC1D24, in whom we identified the same truncating variant in ATP6V1B2 that causes DDOD syndrome (NM_001693.4:c.1516C>T; p.Arg506*). We also studied two families already reported with DDOD syndrome7,8 for whom we performed molecular studies. We thus expand the phenotype associated with ATP6V1B2 variants.
MATERIALS AND METHODS
Ethics statement
This project was approved by the institutional review board of CHU Sainte-Justine. All experiments were performed in accordance with relevant guidelines and regulation. Written informed consent for research was obtained from each participant in the study or their legal representative. Consent for publication of photos included in this article were also obtained.
Participants
Families were identified in a cohort comprising 46 families with DOORS syndrome (based on the presence of three of the five main features forming the acronym) and 2 with DDOD syndrome recruited in a study led by P.M.C. Families in whom no variant in ATP6V1B2 was detected were excluded from this report.
Exome sequencing
Exome sequencing was performed in four individuals with DDOD syndrome and in four with DOORS syndrome, including three previously reported by Campeau et al.1 Sanger sequencing was used to confirm the presence of the variant in other individuals with DOORS syndrome and its absence in healthy relatives.
RESULTS
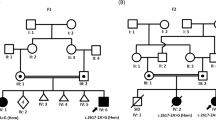
We identified the same truncating variant in ATP6V1B2 (NM_001693.4:c.1516C>T; p.Arg506*) in two families with DDOD syndrome and in nine individuals from eight unrelated families with a clinical diagnosis of DOORS syndrome. The variant was absent in healthy relatives. Table 1 shows other genetic etiologies identified in a total of 46 families assessed ultimately by exome sequencing for DOORS syndrome, including the cohort of 26 families previously reported by Campeau et al.1 The most frequent cause is biallelic variants in TBC1D24, while no genetic cause was identified in 6 families (13%). Our previous publications discuss clinical overlap between DOORS and most conditions mentioned in Table 1.4,9,10,11,12,13 Table 2 and Fig. 1 provide details about phenotypes described in the present report. Figures 2 and 3 show the location of the p.Arg506* variant and previously reported missense variants.

Note onychodystrophy and brachydactyly in all. (a) Individual 4: prognathism, broad nasal ridge, low-set ears, downslanted palpebral fissures, and slight ptosis. (b) Individual 5: broad nasal ridge, downslanted palpebral fissures, and low-set ears. (c) Individual 6: wide base of the nose, downslanted palpebral fissures, long philtrum, and prominent fingerpads. (d) Individual 10: broad nasal ridge and prognathism. (e) Individual 7: bulbous nose, low-set and cupped ears. Radiographs show smaller development and/or delayed ossification of the distal phalanges in both hands and feet.


Image made using PDBe entry 6vq8, the fitted atomic model of cryoEM structure of rat brain V-ATPase complex reported recently.34 The large red spheres are the Arg506 amino acid affected by the nonsense variant (there are three B subunits in the V-ATPase). Smaller red ball and chain amino acids are the amino acids affected by missense variants in other conditions.
Individuals from families 1 and 2 had a diagnosis of DDOD syndrome and typical features of the condition were found in each of them. Additionally, two of them had late dentition. An individual from family 2 also had epilepsy, which was diagnosed at age 36. There was no developmental delay or intellectual disability in either family. No dysmorphisms were observed in family 1. Individuals from family 2 were described with midface under development, deep-set eyes, and hypotelorism, and one of them had two symmetrical areas of aplasia cutis on the vertex of the scalp.
Deafness was present in all individuals with DOORS syndrome, along with onychodystrophy and abnormal fingers and/or toes. Eight individuals presented absent or small nails, while another had pachyonychia and koilonychia. Developmental delay and/or intellectual disability was present in seven individuals, ranging from mild to severe. Individuals from family 3 did not have intellectual impairment. However, the father had an episode of focal seizure at age 39. Four other individuals with DOORS syndrome had seizures. Individual 5 developed tonic–clonic seizures and infantile spasms and individual 6 developed infantile spasms at 12 months of age. Individual 9 presented recurrent tonic–clonic seizures starting at age 52 and individual 10 had tonic–clonic and absence seizures since childhood. Mild dental anomalies, namely late dentition and misalignment, were described in individuals 4 and 5. Some dysmorphic features were shared between individuals. Coarse facies, broad nasal ridge, downslanted palpebral fissures, low-set ears, and prognathism were observed most frequently. Other dysmorphisms include malformed ears, epicanthus, ptosis, high arched palate, long philtrum, prominent fingerpads, thin upper vermilion, synophris, hypertrichosis, and widely spaced teeth. Microcephaly was also present in individual 6. Level of 2-oxoglutarate acid was normal in individuals 4, 5, and 6, and was not assessed in others.
DISCUSSION
We identified a variant in ATP6V1B2 in families with DOORS syndrome. This variant creates a premature stop codon removing five amino acids and results in a truncated protein. The same variant causes DDOD syndrome and has been shown to impair lysosome acidification.2
Clinical findings in our cohort were mostly consistent with previously published patients. Individuals with DDOD syndrome shared sensorineural deafness and onychodystrophy. Regarding digital anomalies, bulbous fingertips of digits, and finger-like thumbs were found in both families, but there was no triphalangeal thumb. Dental anomalies, namely oligodontia and conical teeth, have already been reported,14,15 but those specific features were not observed among the two families we described. However, late dentition was reported in one.
Individuals with DOORS syndrome exhibited typical findings of sensorineural hearing loss, onychodystrophy, osteodystrophy, and different degrees of cognitive impairment. Both triphalangeal thumb and hypoplastic or absent distal phalanges, which are characteristically found in DOORS syndrome,1,16 were observed. Facial dysmorphisms in our cohort also overlapped those previously described, predominantly broad nose. Five of nine had seizures, which are reported in most but not all patients with DOORS syndrome and TBC1D24 variants.1
All previously reported patients with DOORS syndrome had an autosomal recessive inheritance while the ATP6V1B2 variant is associated with an autosomal dominant inheritance, as in DDOD syndrome. Nonetheless, because all families fulfilled clinical criteria for DOORS syndrome instead of DDOD syndrome—with developmental delay or intellectual disability, and seizures as the distinctive features—we considered that they should be designated as having a form of DOORS syndrome.
Variants in TBC1D24 were previously identified in about half of the individuals with DOORS syndrome. TBC1D24 encodes a member of the Tre2-Bub2-Cdc16 (TBC) domain-containing RAB-specific GTPase-activating proteins, which regulate Rab proteins and other GTPases playing a role in the transport of intracellular vesicles.1 TBC1D24 deficiency leads to a defect in neuronal presynaptic endocytosis and an enlarged endosomal compartment resulting in a decreased spontaneous neurotransmission.17 Similarly, TBC1D24 was shown to be critical for removal and degradation of damaged synaptic vesicle proteins from the synapses.18,19 By solving the crystal structure of Sky (fly homolog of TBC1D24), a region of the TBC domain was identified that directly associates with phosphoinositides (PI) in the membrane, especially PI(4,5)P2.20 In addition, certain TBC1D24 variants cause an increased sensitivity to oxidative stress.21 ATP6V1B2, in turn, encodes a component of the V-ATPase, which mediates acidification of eukaryotic intracellular organelles and regulates exocytosis of vesicles. Vacuolar ATPases pump protons in cellular vesicles and have key roles in sensing and regulating cellular pH, vesicular transport, endocytosis, and synaptic vesicle loading.22,23
Previous studies found that cochlear-specific Atp6v1b2-knockdown mice displayed severe sensorineural hearing loss and that Atp6v1b2 c.1516C>T knock-in mice exhibited behavioral defects without hearing loss, suggesting possible hearing compensation mechanisms in the mice and potential cognitive consequences in individuals with DDOD syndrome,24 although intellectual disability has not been associated with DDOD syndrome. Interestingly, TBC1D24-related syndromes also show a wide clinical spectrum, with findings such as intellectual disability not consistent among individuals.25 As such, our new clinical description associating the recurrent ATP6V1B2 variant with DOORS syndrome provides a potentially important missing link between genes known to be associated with overlapping syndromes. TBC1D24 was shown to interact with V-ATPase subunits,26 and it was also the case for other TLDc domain-containing proteins, OXR1 and NCOA7.26,27,28,29 The recurrent variant identified (p.Arg506*) affects an amino acid located in periphery of the protein (Fig. 3), which could suggest a possible impact on protein–protein interactions.
In summary, we described nine individuals with a clinical diagnosis of DOORS syndrome in whom we identified a heterozygous truncating variant in ATP6V1B2. Therefore, we expand the phenotype associated with ATP6V1B2 and propose another causal gene for DOORS syndrome. This finding suggests that DDOD and DOORS syndromes might be on a spectrum instead of being two distinct conditions. Our data provide new clues to help understand the clinical heterogeneity in DOORS syndrome since both proteins are involved in membrane trafficking events, including synaptic vesicle cycling.17,30 Importantly, other members of the V-ATPase complex have been associated with neurodevelopmental disorders causing epilepsy.31,32,33 Further studies are needed to unravel the functional relationship between TBC1D24 and V-ATPase and the impact of their relationship on neurons and cerebral development.
Change history
11 September 2020
In the original version of this Article, the affiliation details for Guillermo Pacheco-Cuellar, Jessica Tardif, Maria Vittoria Camurri, Sirinart Molidperee and Philippe M. Campeau were incorrectly given. They have now been corrected in both the PDF and HTML versions of the Article.
15 September 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41436-020-00969-y
References
Campeau PM, Kasperaviciute D, Lu JT, et al. The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol. 2014;13:44–58.
Yuan Y, Zhang J, Chang Q, et al. De novo mutation in ATP6V1B2 impairs lysosome acidification and causes dominant deafness-onychodystrophy syndrome. Cell Res. 2014;24:1370–1373.
Menendez I, Carranza C, Herrera M, et al. Dominant deafness-onychodystrophy syndrome caused by an ATP6V1B2 mutation. Clin Case Rep. 2017;5:376–379.
Kortum F, Caputo V, Bauer CK, et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann–Laband syndrome. Nat Genet. 2015;47:661–667.
Popp B, Ekici AB, Thiel CT, et al. Exome Pool-Seq in neurodevelopmental disorders. Eur J Hum Genet. 2017;25:1364–1376.
Shaw M, Winczewska-Wiktor A, Badura-Stronka M, et al. EXOME REPORT: novel mutation in ATP6V1B2 segregating with autosomal dominant epilepsy, intellectual disability and mild gingival and nail abnormalities. Eur J Med Genet. 2020;63:103799.
Vind-Kezunovic D, Torring PM. A Danish family with dominant deafness-onychodystrophy syndrome. J Dermatol Case Rep. 2013;7:125–128.
White SM, Fahey M. Report of a further family with dominant deafness-onychodystrophy (DDOD) syndrome. Am J Med Genet A. 2011;155:2512–2515.
Murakami Y, Nguyen TTM, Baratang N, et al. Mutations in PIGB cause an inherited GPI biosynthesis defect with an axonal neuropathy and metabolic abnormality in severe cases. Am J Hum Genet. 2019;105:384–394.
Bauer CK, Schneeberger PE, Kortum F, et al. Gain-of-function mutations in KCNN3 encoding the small-conductance Ca(2+)-activated K(+) channel SK3 cause Zimmermann–Laband syndrome. Am J Hum Genet. 2019;104:1139–1157.
Kariminejad A, Ajeawung NF, Bozorgmehr B, et al. Kaufman oculo-cerebro-facial syndrome in a child with small and absent terminal phalanges and absent nails. J Hum Genet. 2017;62:465–471.
Campeau PM, Hennekam RC, group Dsc. DOORS syndrome: phenotype, genotype and comparison with Coffin–Siris syndrome. Am J Med Genet C Semin Med Genet. 2014;166C:327–332.
Mucha BE, Hennekam RCM, Sisodiya S, Campeau PM. TBC1D24-related disorders. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993.
Robinson GC, Miller JR, Bensimon JR. Familial ectodermal dysplasia with sensori-neural deafness and other anomalies. Pediatrics. 1962;30:797–802.
Kondoh T, Tsuru A, Matsumoto T, Matsuzaka T, Tsuji Y. Autosomal dominant onychodystrophy and congenital sensorineural deafness. J Hum Genet. 1999;44:60–62.
James AW, Miranda SG, Culver K, Hall BD, Golabi M. DOOR syndrome: clinical report, literature review and discussion of natural history. Am J Med Genet A. 2007;143A:2821–2831.
Finelli MJ, Aprile D, Castroflorio E, et al. The epilepsy-associated protein TBC1D24 is required for normal development, survival and vesicle trafficking in mammalian neurons. Hum Mol Genet. 2019;28:584–597.
Fernandes AC, Uytterhoeven V, Kuenen S, et al. Reduced synaptic vesicle protein degradation at lysosomes curbs TBC1D24/sky-induced neurodegeneration. J Cell Biol. 2014;207:453–462.
Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. Loss of Skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell. 2011;145:117–132.
Fischer B, Lüthy K, Paesmans J, et al. Skywalker-TBC1D24 has a lipid-binding pocket mutated in epilepsy and required for synaptic function. Nat Struct Mol Biol. 2016;23:965–973.
Luthy K, Mei D, Fischer B, et al. TBC1D24-TLDc-related epilepsy exercise-induced dystonia: rescue by antioxidants in a disease model. Brain. 2019;142:2319–2335.
Vavassori S, Mayer A. A new life for an old pump: V-ATPase and neurotransmitter release. J Cell Biol. 2014;205:7–9.
Bell S, Rousseau J, Peng H, et al. Mutations in ACTL6B cause neurodevelopmental deficits and epilepsy and lead to loss of dendrites in human neurons. Am J Hum Genet. 2019;104:815–834.
Zhao W, Gao X, Qiu S, et al. A subunit of V-ATPases, ATP6V1B2, underlies the pathology of intellectual disability. EBioMedicine. 2019;45:408–421.
Kimble JE, White JG. On the control of germ cell development in Caenorhabditis elegans. Dev Biol. 1981;81:208–219.
Merkulova M, Paunescu TG, Azroyan A, Marshansky V, Breton S, Brown D. Mapping the H(+) (V)-ATPase interactome: identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci Rep. 2015;5:14827.
Merkulova M, Paunescu TG, Nair AV, et al. Targeted deletion of the Ncoa7 gene results in incomplete distal renal tubular acidosis in mice. Am J Physiol Renal Physiol. 2018;315:F173–F185.
Huttlin EL, Ting L, Bruckner RJ, et al. The BioPlex Network: a systematic exploration of the human interactome. Cell. 2015;162:425–440.
Doyle T, Moncorge O, Bonaventure B, et al. The interferon-inducible isoform of NCOA7 inhibits endosome-mediated viral entry. Nat Microbiol. 2018;3:1369–1376.
Marshansky V, Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol. 2008;20:415–426.
Fassio A, Esposito A, Kato M, et al. De novo mutations of the ATP6V1A gene cause developmental encephalopathy with epilepsy. Brain. 2018;141:1703–1718.
Mucha BE, Banka S, Ajeawung NF, et al. A new microdeletion syndrome involving TBC1D24, ATP6V0C, and PDPK1 causes epilepsy, microcephaly, and developmental delay. Genet Med. 2019;21:1058–1064.
Van Damme T, Gardeitchik T, Mohamed M, et al. Mutations in ATP6V1E1 or ATP6V1A cause autosomal-recessive cutis laxa. Am J Hum Genet. 2017;100:216–227.
Abbas YM, Wu D, Bueler SA, Robinson CV, Rubinstein JL. Structure of V-ATPase from the mammalian brain. Science. 2020;367:1240–1246.
Acknowledgements
We thank all families. This study was funded in part by Canadian Institutes of Health Research and Fonds de la Recherche du Québec–Santé awards to P.M.C.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Beauregard-Lacroix, E., Pacheco-Cuellar, G., Ajeawung, N.F. et al. DOORS syndrome and a recurrent truncating ATP6V1B2 variant. Genet Med 23, 149–154 (2021). https://doi.org/10.1038/s41436-020-00950-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-00950-9
Keywords
This article is cited by
-
Genetic architecture and phenotypic landscape of deafness and onychodystrophy syndromes
Human Genetics (2022)
-
Correspondence on “DOORS syndrome and a recurrent truncating ATP6V1B2 variant” by Beauregard-Lacroix et al.
Genetics in Medicine (2021)
-
Response to Gao et al.
Genetics in Medicine (2021)
