
Abstract
Purpose
Clinical data provided to genetic testing laboratories are frequently scarce. Our purpose was to evaluate clinical scenarios where phenotypic refinement in proband’s family members might impact exome data interpretation.
Methods
Of 614 exomes, 209 were diagnostic and included in this study. Phenotypic information was gathered by the variant interpretation team from genetic counseling letters and images. If a discrepancy between reported clinical findings and presumably disease-causing variant segregation was observed, referring clinicians were contacted for phenotypic clarification.
Results
In 16/209 (7.7%) cases, phenotypic refinement was important due to (1) lack of cosegregation of disease-causing variant with the reported phenotype; (2) identification of different disorders with overlapping symptoms in the same family; (3) similar features in proband and family members, but molecular cause identified in proband only; and (4) previously unrecognized maternal condition causative of child’s phenotype. As a result of phenotypic clarification, in 12/16 (75%) cases definition of affected versus unaffected status in one of the family members has changed, and in one case variant classification has changed.
Conclusion
Detailed description of phenotypes in family members including differences in clinical presentations, even if subtle, are important in exome interpretation and should be communicated to the variant interpretation team.
Similar content being viewed by others


INTRODUCTION
Identification of disease-causing variants in exome sequencing (ES) data remains an ongoing challenge. Potential analytical causes of missed or misinterpreted variants include wrong transcript selection,1 mosaicism,2 and lack of inclusion of information on gene–disease correlation in the databases,3 and incomplete communication or understanding of the phenotype.3 The American College of Medical Genetics and Genomics (ACMG) variant classification system mostly deals with molecular and functional aspects of variants. The criteria mentioning phenotype include PS4 (“consistent phenotypes”), PP4 (“phenotypes specific for disease with single etiology”), and PP1/BS4 (“segregation or lack of segregation of candidate variant with disease phenotype”). In addition, PS2/PM6 criteria for de novo variants can be applied if “the phenotype in the patient matches the gene’s disease association with reasonable specificity.”4
Clinical data provided to genomic laboratories are frequently scarce. To understand if phenotypic presentation is compatible with molecular findings, in-depth phenotypic information is sometimes required. Requirements for a minimal set of phenotypic features to be communicated to laboratories have not yet been defined.
Our purpose was to describe in which variant interpretation scenarios phenotypic refinement might be important in exome data interpretation.
MATERIALS AND METHODS
Ethics statement
The study was approved by the Rabin Medical Center (RMC) institutional review board committee. Patients’ consent for this study was received and archived.
Study setting and participants
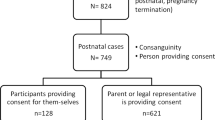
The study cohort consists of ES tests that were interpreted at the Raphael Recanati Genetics Institute, RMC. Individuals presenting with multiple congenital abnormalities, dysmorphic features, global developmental delay/intellectual disability or abnormalities of organ systems with possible heterogeneous etiology were referred for ES by clinical geneticists following normal chromosomal microarray and, when appropriate, single-gene or panel testing. During 2015–2020, 614 tests were performed; 209 were diagnostic. All participants were evaluated by a clinical geneticist. ES trio was performed in 76.5% of the cases, only one parent (with or without additional siblings) was tested in 20.6% of cases, and the proband only was tested in 2.9% of the cases. For ES data analysis, phenotypic information was collected by an in-house variant interpretation team from referral letters and images of patients and, when available, parents. If there was a discrepancy between reported clinical findings and presumably causative variant segregation, referring clinicians were contacted for phenotypic clarification. If needed, the families were asked by clinicians to provide additional phenotypic information. Cases where there was a discrepancy between reported clinical findings and presumably disease-causing variant segregation are described in detail in this report.
Exome sequencing
Of 614 cases, 162 were sequenced by external laboratories, and ES data were interpreted both by external laboratories and departmental team, as described.3 An additional 127 cases were sequenced as part of the research collaboration between RMC and Regeneron, as described.5 Finally, 325 cases were sequenced by the CeGat laboratory (CeGaT GmbH, Tuebingen, Germany) as a clinical service. Targeted capture of protein-coding regions was performed using SureSelectXT Exome V6 (Agilent Technologies, Santa Clara, CA, USA) or Twist Human Core Exome or Exome Plus Kit (Twist Bioscience, San Francisco, CA, USA). Paired-end libraries were sequenced on Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA); at least 97% of target bases were covered at 20× or greater (95% at >100×).
Bioinformatics
The FASTQ files and phenotypic information using Human Phenotype Ontology (HPO) terms were uploaded into Emedgene’s platform (Emedgene Technologies, Mazor, Israel) and analyzed as described previously.3 Filtration parameters included variant quality (mapping quality ≥45 and depth ≥10), population frequency (1% or 5% for dominant or recessive inheritance, respectively) and variant impact on the protein. In addition, autoanalysis identified ~10 variants likely to solve the case. Variants were classified according to ACMG criteria.4 Analysis of copy-number variants was not performed.
Segregation analysis
If segregation in additional family members was needed, variants of interest were analyzed by Sanger sequencing.
RESULTS
A diagnosis was determined in 209/614 ES cases (34%). Disease-causing variants in 216 genes having known gene–disease associations were identified. Tables S1 and S2 summarize characteristics of the diagnostic cohort and modes of inheritance in the diagnosed cases. In an additional ten cases, a strong candidate variant in a gene with no previously known gene–disease association was identified by the team; seven of these have been subsequently published.6,7,8,9,10,11,12 These cases were excluded from the study.
Table 1 summarizes 16 cases, grouped into four categories, where the discrepancy between candidate variant segregation and expected disease status in one of the family members (affected versus unaffected) necessitated refinement of the phenotypic information.
Proband and parent carried the same causative variant, but the parent was not reported as clinically affected
In this category we encountered 11/209 cases (5.3%). As an example, in case 1, the mother carrying an NFIA variant causing brain malformations with or without urinary tract defects was considered to be clinically affected only following phenotypic clarification. Similarly, in cases 2, 4, and 5, the transmitting parent was not reported as affected but inspection of the facial photographs led to the conclusion that these parents are affected. In cases where following phenotypic clarification the transmitting parent was still considered unaffected, either nonpenetrance was known for a disorder diagnosed (cases 8 and 9) or parental mosaicism has been observed (cases 10 and 11). In total, in seven cases phenotypic clarification led to a change in clinical definition of parental disease status (cases 1–7).
Different disorders with overlapping symptoms in proband and/or parents
In this category 2/209 (0.96%) cases were observed. In case 12, two different disorders in the same family were characterized by macrocephaly, which was borderline in the mother carrying ACAN variant, but extreme in the proband with disease-causing variants in both ACAN and TAOK1. Severe macrocephaly of +3.8 standard deviation in the child was caused by the coexistence of both conditions. In case 13, two different disorders in different family members with a similar phenotype were identified; short stature in the proband was caused by de novo IGF1R variant causing insulin-like growth factor I resistance, while short stature in the father was caused by homozygous recessive MMP13 variant related to Missouri type spondyloepimetaphyseal dysplasia. In addition to short stature, the proband had microcephaly related to an IGFR1 variant, while paternal head circumference was normal.
Similar phenotypes in the proband and in other family members, but molecular cause identified only in the proband
In this category there were 2/209 cases (0.96%). In case 14, two fetuses with increased nuchal translucency and short limbs on the fetal ultrasound underwent ES, but a de novo SOX9 pathogenic variant causing campomelic dysplasia with autosomal sex reversal was identified in only one fetus. Clinical follow-up of the second fetus showed ultrasonic resolution of the previously suspected limb abnormalities. In case 15, both the proband and the mother were reported to have renal cysts. ES analysis revealed pathogenic PKD1 variant in the proband, but not in the mother. Acquisition of additional phenotypic information revealed that the mother had only a few unilateral renal cysts (identified as a result of proband’s phenotype), while the proband had multiple bilateral renal cysts. Therefore, maternal disease status could be changed to unaffected.
Previously unknown maternal condition was identified as causative of child’s phenotype
In this category there were 1/209 cases (0.5%). In case 16, due to identification of homozygous pathogenic variant in PAH in the mother, maternal hyperphenylaninemia was suspected to be causative of proband’s phenotype. The clinician was contacted and previously unknown maternal hyperphenylalaninemia has been biochemically confirmed. In this case, phenotypic clarification has led to a change in both mother’s and proband’s disease status.
In summary, in 16/209 (7.7%) cases, due to a discrepancy between the molecular findings and disease status assignment in parents or siblings, it was considered important by the variant interpretation team to further clarify phenotypic information with the referring clinician. In total, in 12/16 (75.0%) cases disease status of one of the family members has changed from unaffected to affected or vice versa—in 11 cases parental and in 1 case sibling’s disease status assignment has changed. However, in only one case (case 2) this led to a change in variant classification, from variant of unknown significance to likely pathogenic (by removing BS4 criterion).
DISCUSSION
We report examples where refinement of phenotypic information on parents or siblings of the proband led to a change in clinical family members’ disease status assignment in accordance to the segregation of the causative variant discovered on ES. This led to more confidence that a variant is indeed causative of patients’ phenotype. However, only in one case did phenotypic refinement lead to a change in variant classification accordingly to ACMG criteria. Interaction between the variant interpretation team and referring clinician was especially important for dominant genes, in situations where the transmitting parent was not clearly defined as affected according to the clinical geneticist’s summary letter. Observing an unaffected transmitting parent would be unusual for disorders where incomplete penetrance has not been reported, unless an individual is mosaic for the causative variant. In addition, previously reported information on penetrance might be incomplete if the disorder is very rare or only recently described. It turned out that, in many cases, a transmitting parent showed phenotypic features, but he or she was not clearly described as affected in the summary letter. More mothers (5) than fathers (1) with underreported mild cognitive abnormality and/or mild dysmorphism were observed. This is not surprising since some developmental disorders show reduced penetrance and expressivity in females compared with males.13 In some disorders, e.g., KBG syndrome, it is known that a mildly affected mother can be diagnosed after a typically affected son is recognized.14 In the case of KBG syndrome in our cohort, it was the transmitting father who was diagnosed as having this disorder following exome trio analysis. Moreover, the proband had no dysmorphic features characteristic of KBG syndrome, while the father had typical dysmorphic features not reported in the summary letter. We suspect that, in many cases, the reason for underreported parental cognitive difficulties might be physician reluctance (due to psychological discomfort) to provide a family with a summary letter stating that one of the parents is cognitively abnormal. In addition, in certain situations it is not possible to unequivocally estimate parental cognitive status during child’s visit, since parental medical documentation is unavailable or one of the parents is not present during the visit and arrives at the clinic for ES testing only. Because a disease-causing variant might be missed due to incorrectly reported disease status, especially dominant missense variant, it is important to invite both parents to the clinic and to carefully document parental phenotypic status.
A less common but important scenario was identification of a presumably fully penetrant variant in mosaic state in a phenotypically normal parent. Mosaicism in autosomal dominant disorders is a well-known phenomenon.2 When mosaicism is present, the bioinformatics variant analysis platform might automatically mark an individual as either heterozygous or wild type, depending on variant filtering settings and on number of reads carrying the disease-causing variant. From the clinical point of view, the phenotype of a mosaic individual might be normal, relatively mild, or full-blown. As exemplified by case 10, a mosaic asymptomatic mother with a variant in the DDX3X gene causing a dominant disorder (mental retardation, X-linked 102) gave birth to three affected daughters.
In the category of double diagnosis, also a well-known phenomenon,15 particularly confusing were situations when different rare disorders with overlapping clinical features presented in the proband and the parents.
Reviewing images of the proband and the parents to correctly assign parental disease status following identification of disease-causing variant was important in three cases in our cohort.
In conclusion, by showing situations where a causative variant might be missed or misclassified due to mistaken assignment of the disease status of the individual tested, we emphasize that phenotypic information should be collected and communicated to the variant interpretation team not only for the proband but also for family members. Categories of potentially helpful phenotypic information are listed in Table S3. Providing variant interpretation teams with a thorough description of parental cognitive status and physical findings, as well as collecting parental photos, including their photos from childhood, are of utmost importance. In addition, differences in clinical presentations in family members, even if subtle, should be clearly documented. We think that inclusion of clinical geneticists into ES data interpretation teams will help to recognize situations where additional phenotypic data should be obtained and matched with molecular data.
References
Wai HA, Lord J, Lyon M, et al. Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genet Med. 2020;22:1005–1014.
Cao Y, Tokita MJ, Chen ES, et al. A clinical survey of mosaic single nucleotide variants in disease-causing genes detected by exome sequencing. Genome Med. 2019;11:48.
Basel-Salmon L, Orenstein N, Markus-Bustani K, et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet Med. 2019;21:1443–1451.
Richards S, Aziz N, Bale S, ACMG Laboratory Quality Assurance Committee, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Bend R, Cohen L, Carter MT, Lyons MJ, et al. Phenotype and mutation expansion of the PTPN23 associated disorder characterized by neurodevelopmental delay and structural brain abnormalities. Eur J Hum Genet. 2020;28:76–87.
Tan TY, Sedmík J, Fitzgerald MP, et al. Bi-allelic ADARB1 variants associated with microcephaly, intellectual disability, and seizures. Am J Hum Genet. 2020;106:467–483.
Mak CCY, Doherty D, Lin AE, et al. MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis. Brain. 2020;143:55–68.
Li L, Ghorbani M, Weisz-Hubshman M, et al. Lysine acetyltransferase 8 is involved in cerebral development and syndromic intellectual disability. J Clin Invest. 2020;130:1431–1445.
Horn S, Au M, Basel-Salmon L, et al. De novo variants in PAK1 lead to intellectual disability with macrocephaly and seizures. Brain. 2019;142:3351–3359.
Magini P, Smits DJ, Vandervore L, et al. Loss of SMPD4 causes a developmental disorder characterized by microcephaly and congenital arthrogryposis. Am J Hum Genet. 2019;105:689–705.
Orenstein N, Goldberg-Stern H, Straussberg R, et al. A de novo GABRA2 missense mutation in severe early-onset epileptic encephalopathy with a choreiform movement disorder. Eur J Paediatr Neurol. 2018;22:516–524.
Weisz Hubshman M, Broekman S, van Wijk E, et al. Whole-exome sequencing reveals POC5 as a novel gene associated with autosomal recessive retinitis pigmentosa. Hum Mol Genet. 2018;27:614–624.
Tropeano M, Ahn JW, Dobson RJ, et al. Male-biased autosomal effect of 16p13.11 copy number variation in neurodevelopmental disorders. PLoS One. 2013;8:e61365.
Brancati F, Sarkozy A, Dallapiccola B. KBG syndrome. Orphanet J Rare Dis. 2006;1:50.
Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31.
Waseem NH, Vaclavik V, Webster A, Jenkins SA, Bird AC, Bhattacharya SS. Mutations in the gene coding for the pre-mRNA splicing factor, PRPF31, in patients with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2007;48:1330–1334.
Rice GI, Kasher PR, Forte GM, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248.
Guo H, Bettella E, Marcogliese PC, et al. Disruptive mutations in TANC2 define a neurodevelopmental syndrome associated with psychiatric disorders. Nat Commun. 2019;10:4679.
Dulovic-Mahlow M, Trinh J, Kandaswamy KK, et al. De novo variants in TAOK1 cause neurodevelopmental disorders. Am J Hum Genet. 2019;105:213–220.
Acknowledgements
The manuscript was edited by Debby Mir.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
CG-J and ARS are full-time employees of the Regeneron Genetics Center from Regeneron Pharmaceuticals Inc. and receive stock options as part of compensation. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Basel-Salmon, L., Ruhrman-Shahar, N., Orenstein, N. et al. When phenotype does not match genotype: importance of “real-time” refining of phenotypic information for exome data interpretation. Genet Med 23, 215–221 (2021). https://doi.org/10.1038/s41436-020-00938-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-00938-5
Keywords
This article is cited by
-
Heterozygous truncating variant of TAOK1 in a boy with periventricular nodular heterotopia: a case report and literature review of TAOK1-related neurodevelopmental disorders
BMC Medical Genomics (2024)
-
Phenotypic compatibility and specificity in genomic variant classification
European Journal of Human Genetics (2024)
-
Ordering genetic testing by neurologists: points to consider
Journal of Neurology (2023)
-
Application of exome sequencing for prenatal diagnosis of fetal structural anomalies: clinical experience and lessons learned from a cohort of 1618 fetuses
Genome Medicine (2022)
