
Abstract
Purpose
Genomic newborn screening raises practical and ethical issues. Evidence is required to build a framework to introduce this technology safely and effectively. We investigated the choices made by a diverse group of parents with newborns when offered tiered genomic information from exome sequencing.
Methods
This population-derived cohort comprised infants with congenital deafness. Parents were offered exome sequencing and choice regarding the scope of analysis. Options were choice A, diagnostic analysis only; choice B, diagnostic analysis plus childhood-onset diseases with medical actionability; or choice C, diagnostic analysis plus childhood-onset diseases with or without medical actionability.
Results
Of the 106 participants, 72 (68%) consented to receive additional findings with 29 (27.4%) selecting choice B and 43 (40.6%) opting for choice C. Family size, ethnicity, and age of infant at time of recruitment were the significant predictors of choice. Parents who opted to have additional findings analysis demonstrated less anxiety and decisional conflict.
Conclusions
These data provide evidence from a culturally diverse population that choice around additional findings is important and the age of the infant when this choice is offered impacts on their decision. We found no evidence that offering different levels of genomic information to parents of newborns has a negative psychological impact.
Similar content being viewed by others


INTRODUCTION
Genomic sequencing has become the gold standard for the investigation and diagnosis of monogenic diseases.1 Sequencing generates a large amount of “off target” data that may be relevant to future health and there is significant variability in how these data are managed.2 Recommendations have been developed for reporting additional or secondary findings in a defined list of genes3 in an attempt to standardize practice. With the ability to understand and interpret this genetic information, there is emerging potential for its use as a public health tool. An additional layer of complexity exists when the tested individual is a child.4,5
As the use of genomic tests for disease prediction and precision medicine is realized, genomic newborn screening becomes a possibility. There is a dichotomy of opinions around public health genomics and genomic newborn screening (NBS). One view recommends liberal testing and augmentation of current NBS protocols with genomics, reasoning that the information is wanted by families6,7 and is of benefit to screened individuals.8 Significant research is underway to investigate and provide evidence to underpin this approach.9 A more conservative view is that the complexity of consent, cost, and interpretation are still significant barriers to genomic newborn screening and that the public health benefit has not been rigorously demonstrated.10,11 In addition, there is a gap in the knowledge around what parents with newborns want out of genomic testing and what impact genomic newborn screening could have when it is offered as a test rather than as a hypothetical concept.
Current practice in Australia is for all newborns to have bloodspot screening for 24 serious and treatable diseases.12 Additionally, there is state-based newborn hearing screening that covers all birthing hospitals.13 Here we describe the Baby Beyond Hearing project, offering parents who have already consented to exome sequencing to investigate the cause of their child’s hearing impairment a choice with respect to the amount and type of additional genetic information they wished to receive. The aims were to investigate the choices made by parents and the potential predictors of choice when different levels of genomic information from exome sequencing are offered, and to determine if there was any associated adverse psychosocial impact.
MATERIALS AND METHODS
The methods are described in detail in the protocol publication14 and are summarized below. This study was part of the Melbourne Genomics Health Alliance program and received Human Research Ethics Committee approval from Melbourne Health, 2013.245. Informed written consent was obtained from all participants.
Study design and participants
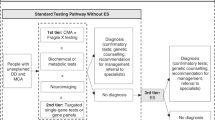
Infants born in the state of Victoria, Australia in 2016 or 2017 and diagnosed with bilateral moderate, severe, or profound hearing impairment following newborn hearing screening were eligible to participate. Participants were recruited during clinical care at pediatrician-run deafness clinics at the Royal Children’s Hospital and Monash Children’s Hospitals in Melbourne, as well as in regional genetics outreach clinics across the state. The deafness clinics had different models of care with one offering a single visit service following diagnosis of congenital deafness and the other running as an ongoing management clinic, which affected waiting times and therefore age at recruitment. Sample size was opportunistic, based on how many babies were born in the two-year time frame who fitted the study inclusion criteria for the primary project investigating the genetic causes of congenital deafness.15 All participants received a decision support tool and genetic counseling. If parents consented to exome sequencing to investigate the cause of their child’s hearing impairment, they were offered three choices, as outlined in Fig. 1 and explained further below.

Choices presented for parents receiving results from exome sequencing in their infants with congenital hearing impairment.
Decision support tool
A decision support tool (see Supplementary S1) was modeled from one developed for prenatal diagnostic testing using chromosome microarrays, and offering different levels of genetic information.16 It explained exome sequencing, the possible outcomes of having diagnostic testing, and the option of receiving additional findings in addition to diagnostic information. Worksheets were included to guide discussion and help parents weigh the personal advantages and disadvantages of receiving different levels of information. The decision support tool was provided to parents by post or email prior to a genetic counseling session. Both parents were encouraged to read and consider the information together, particularly if only one parent was available to attend face-to-face genetic counseling.
Genetic counseling
All genetic counselors attended a one-day workshop conducted by senior genetic counselors. The workshop focused on the differences between counseling for a medically indicated clinical test versus that for additional findings. Role plays were designed to allow counselors to practice potential hurdles that may be faced in these clinical encounters, for example, a couple that had different views about how much information they wanted for their child.
Exome sequencing, gene list analysis, and reporting
Exome sequencing was performed at the Victorian Clinical Genetics Services clinically accredited laboratory. Participants had analysis of genes according to the consent provided as described below. Standard practice in the laboratory is to mask all variants not selected for analysis. Gene lists offered to parents for analysis are summarized in Fig. 1 and provided in full in Table S2. Choice A was analysis of a deafness gene list15 with expanded analysis if there was a clinical indication as determined by the clinical geneticist. Choice B comprised choice A genes plus additional analysis of genes associated with conditions that have onset in childhood that are considered medically actionable.17 Choice C was choice B plus additional analysis of genes that were not necessarily medically actionable but had definitive gene disease association,18 onset of symptoms prior to the age of 16 years the majority of the time, and a validation method available, such as biochemical test or clinical examination, to confirm diagnosis at the time of testing. The validation method was included to minimize potential harms from providing predictive genetic information that included a large degree of uncertainty. After genetic counseling, parents provided written informed consent for testing, but could change their choice of gene list analysis at any time during the study period if the additional findings report had not been issued. Only one parent was required to sign the consent; however, joint decision-making was strongly encouraged. Counselors offered a follow-up phone call if the family felt undecided at the time of appointment or if a parent had not been present for the session and required further consultation.
Variants were classified according to the principles outlined in the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) standards for interpretation of sequence variants.19 Variant classification was reviewed in a multidisciplinary team meeting attended by clinical geneticists, medical subspecialists, genetic counselors, medical scientists, and bioinformaticians. Only variants classified as pathogenic or likely pathogenic were reported for choice B and choice C analysis. If a pathogenic or likely pathogenic variant for an autosomal recessive condition was reported, a variant of uncertain significance (VUS) in the same gene was reportable. This component of analysis was conducted separately from diagnostic analysis and a separate report was issued. This was done intentionally to allow differences in diagnostic versus predictive analysis to be outlined. Results of the diagnostic analysis of deafness genes in the cohort are reported elsewhere.15
Return of results
Return of diagnostic results (choice A) was at a face-to-face appointment with a clinical geneticist and genetic counselor. If results of additional findings analysis were available, these were provided simultaneously. If not available, these results were provided either over the phone (negative results only) or at a face-to-face appointment. Where possible, the face-to-face appointment was incorporated into the pediatrician follow up at the deafness clinic.
Outcomes
The primary outcome measure was the proportion of participants choosing each of the three analysis options. The secondary measures were predictors of choice and the psychological impact of the process on parents of infants. Data collection and surveys were administered at four timepoints (Fig. 2).

Flowchart of patient recruitment and data collection timepoints.
Data collection
Families were asked to complete a questionnaire for data collection prior to or during the first clinic visit. Surveys were also provided to families after consent for testing, after receiving results for diagnostic testing, and after receiving results for additional findings (Fig. 2). The surveys included questions on demographics such as postcode, parental education and family income and scales to measure medical literacy,20 anxiety,21 decisional conflict and regret,22,23 and intolerance of uncertainty.24
Data analysis
Survey data were analyzed using Stata version 14 (StataCorp LLC, TX, USA). Comparisons between the three outcome groups, representing choice of analysis, were undertaken using chi2 tests for categorical variables and analysis of variance (ANOVA) or independent t tests for normally distributed continuous predictor variables.
Multivariable analyses to determine the independent predictors of choice were undertaken using logistic regression. Variables with p values ≤0.2 in the univariable analysis were included in the adjusted model to account for their possible confounding effect. Results were reported as odds ratios (ORs) and adjusted OR with 95% confidence intervals (CIs) and p values.
RESULTS
One hundred seventy (170) patients were identified as eligible for the project, of whom 156 were able to be contacted and invited to participate. Of the 106 participants who consented to having diagnostic exome sequencing for their child’s hearing impairment, 72 (68%) consented to receiving additional findings with 29 (27.4%) opting for choice B (medically actionable childhood-onset conditions) and 43 (40.6%) opting for choice C (including non–medically actionable childhood-onset conditions).
The decision support tool was utilized by 63/106 (59%) of participants. Thirty families (28%) had made a choice prior to the genetic counseling appointment but eight of these changed their choice following a face-to-face genetic counseling session. Fifteen families (14%) changed their decision at some time after the face-to-face appointment: ten families signed consent for choice A and then opted into additional findings, three families opted out of additional findings following initial consent, and two families altered the amount of additional findings information they chose to receive (both went from choice B to C). One family declined to receive the results of additional findings after the report had been issued.
Data collection
The initial data collection questionnaire was completed in clinic and therefore had 100% response rate. Survey 1, administered after consent to participate and choice of A, B, or C gene list analysis was made, was completed by 63/106 families. Survey 2, which was taken after results for diagnostic analysis were provided, was completed by 52/106. For those that opted in to receive additional findings, 47/72 completed survey 3 after results were disclosed.
Participant characteristics within each choice group
Characteristics of the 106 participants were collected in the initial questionnaire and are summarized in Table 1. Table 1 shows differences in characteristics between those who chose A, B, or C and between those who chose A (declined additional findings) versus those who chose either B or C (consented to additional findings). There was no difference in the mean maternal or paternal age, sex of the baby, use of assisted reproduction, or gestation at delivery between groups. The majority of families were recruited when their infants were between 3 and 9 months of age. In the families who were approached before their infant was 3 months old, there was a lower uptake of additional findings. Those with two children showed a tendency toward seeking medically actionable additional findings (choice B) compared with first-time parents or those with larger families. Parents who stated their child was Australian or New Zealander were more likely to request additional genomic information than all other ethnic groups. If the infant had a medical problem in addition to hearing impairment, there was a preference for parents to opt for all available information (choice C). Use of the decision support tool was higher in those who opted for additional findings, with 64% of those who chose additional findings (choice B/C) using the tool compared with 50% of those who opted for diagnostic analysis only (choice A). Additional sociodemographic information was collected in survey 1, which was completed by 63 parents of participants. These data are presented in Table 1. Education, medical literacy, and family income were not different between groups.
Predictors of choice: multivariable analysis
Table 2 shows results of univariable and multivariable analyses, comparing those who declined additional findings (choice A) with those who consented to additional findings (choices B and C combined). Covariates included in all multivariable analyses were those with a p value ≤0.2 in either ANOVA ort test: family size, ethnicity of child, age at recruitment, other medical problems, and use of the decision aid. There was an almost fivefold increased likelihood that there would be a choice of additional findings in families with two children, compared with those with one child. Likewise, when parents identified their child as Australian or New Zealander they were four times more likely to choose additional findings compared with other identified ethnicities for the child, no matter whether adjustments for covariates were made or not. Families approached prior to the infant being 3 months of age compared with those with infants greater than 3 months were markedly less likely to consent to additional findings (adjusted OR 0.06, 95% CI: 0.01–0.35, p = 0.002), again independent of adjusting for the covariates.
Impact of choice on psychosocial measures
State anxiety, decisional conflict, and intolerance of uncertainty were measured in survey 1. Table 3 shows that participants who sought additional findings had less anxiety at the time of decision-making, less conflict around their decision, and were more tolerant of uncertainty. Decisional regret was low across all groups.
Reportable findings
From the choice A gene list (diagnostic analysis), 56% of participants received a genetic diagnosis, the details of which are published elsewhere.15 Four infants had reportable variants identified in the additional findings analysis. All reported variants were in genes in the choice B gene list (actionable childhood-onset conditions). Two of these participants required further investigation and follow up.
Two participants had a heterozygous variant in MEFV c.228G>A classified as pathogenic.MEFV is associated with familial Mediterranean fever (FMF), which is generally considered an autosomal recessive condition; however, some heterozygotes show an incomplete or atypical form of the disease.25 For this reason, variants in this gene were considered for both dominant and recessive inheritance and heterozygous likely pathogenic or pathogenic variants were reported. The c.228G>A variant has not been associated with autosomal dominant inheritance in the literature and the families were counseled that the result was likely to represent carrier status, although they were informed about the symptoms and signs of FMF. No further investigations or follow up were organized.
In one participant, two variants were identified in the APOB gene: c.10190del, (pathogenic) and c.12635C>G (variant of uncertain significance [VUS]). Two allelic diseases can occur due to variants in APOB: familial hypercholesterolemia is the result of missense variants and results in high levels of high density lipoprotein and cholesterol predisposing the individual to early and severe coronary artery disease,26 whereas truncating variants cause hypobetalipoprotinemia with resultant reduced absorption of fat soluble vitamins, failure to thrive, and vitamin deficiencies.27 This participant had normal cholesterol and fat soluble vitamin levels, and no further referral or follow up was planned.
In another participant, a heterozygous variant was reported in theVWF gene, c.7390C>T (pathogenic), which has been associated with von Willebrand disease. There was a history of easy bruising in the proband and a family history of a sibling and maternal grandmother with frequent nosebleeds. The infant had von Willebrand factor testing, which was in the low normal range. The family is awaiting further hematology assessment. Parents sought cascade testing for themselves and four other children.
DISCUSSION
While evidence is mounting for the utility and cost effectiveness of genomic sequencing in the newborn period for diagnostic purposes28,29 there is still limited knowledge of the scope of findings that should be offered and reported to parents in a real-world setting. The Baby Beyond Hearing project provides insight into parents’ choices, predictors of these choices, and the psychosocial impact of offering additional findings from genomic sequencing to parents of newborns. Overall, approximately two-thirds of parents elected to receive additional information from their infant’s exome sequencing, but a sizable 32% did not want to receive additional findings. Of particular relevance to newborn screening was the finding that parents with an infant less than 3 months of age were the least likely to elect to receive additional findings compared with parents with infants older than 3 months.
Our results highlight that people seek different levels of information from genomic sequencing according to their personal values and circumstances. Our findings are consistent with quantitative and qualitative studies offering parents hypothetical choices in relation to genomic newborn screening in which there is no consensus on how much information people seek.30 The approximately 70:30 ratio of seeking broader, predictive genetic information versus targeted information is mirrored in other studies of parents having genetic testing prenatally or for their newborn, despite variations in study design and population.16,31,32
The lower rate of uptake by parents to seek predictive genomic information when their infant was less than 3 months old has been replicated in other research32 and may be a significant barrier when considering using this technology to complement or expand traditional newborn screening. It is possible this is confounded by a disinclination of new parents to participate in research generally. A further practical barrier to public health implementation of genomic newborn screening is the fragility of parents’ decision-making. The high rate of participants who changed their decision over the the study time period emphasizes the lack of assurance in the decision and the importance of ongoing genetic counselor support that provides flexible consent.
Qualitative studies in a hypothetical setting suggest individualized support is required for people to make a choice about genomic testing for their infants33 and highlight the important impact of social context and cultural difference in making these choices. A strength of this study is the diversity of the population included. Having access to a population cohort and interpreter services makes these data unique among research into newborn genomic screening, which often reflects a more limited population subset.32 Cultural orientation, which influences life experiences, has an impact on desires and expectations around additional findings.33 Our results support this premise in a real-world setting, with those identifying as Australian or New Zealander being much more likely to seek additional genomic information than all other ethnic groups. This may reflect greater confidence in the health-care system or a higher value and understanding placed on Western medicine.
Similarly, family size impacts on parents’ decision-making, and this may be linked to social context and cultural factors.34 One possibility is that a smaller and therefore “younger” family may seek broader genetic information to inform further reproductive decisions. This will be explored further in planned qualitative analysis.
These results provide the first published quantitative data on the psychosocial impact of offering genomic newborn screening to parents. Data from similar projects are likely to follow35,36 and will provide further insight into cultural and social preferences across continents. While hypothetical surveys have demonstrated concern from health professionals about the psychosocial impact on families,37 our results demonstrate overall low levels of anxiety and feelings of conflict and regret around the decision to have genomic newborn screening. Participants who sought additional findings had significantly less anxiety at the time of decision-making, less conflict around their decision, and were more tolerant of uncertainty. Decisional regret scores were very low among all groups, reflecting that people were comfortable with the decisions they made, although survey completion rates fell and therefore these data are incomplete. These findings could reflect the optimism parents feel around having newborn genomic screening for childhood-onset conditions38 and appear to contrast with the ambivalence described in studies offering parents screening of their children for adult-onset treatable conditions.39 Qualitative analysis of surveys and participant interviews are underway and this will provide further insight into the psychosocial impact and personal utility of offering genomic newborn screening and essential information about longer-term impact of these decisions for parents.
The scope of gene lists analyzed may influence parental choices and should be carefully considered when offering predictive genomic information. Increased availability of sequencing data in healthy individuals will strengthen understanding of penetrance and improve the robustness of selection of genes for inclusion in the future. With incomplete penetrance data we deemed it appropriate to provide a second validation method for genes included for choice C. This reduced the potential for harm that could arise from giving parents information with a high degree of uncertainty about their child's future health. It is possible that this made parents more likely to select choice C; however, our results indicate that these individuals were more tolerant of uncertainty and therefore may have made this choice without a validation method available.
A limitation of this study in terms of generalizability is that this cohort was derived from a population of hearing impaired infants and therefore results cannot necessarily inform predictive genomic sequencing on healthy newborns. Likewise it is not necessarily applicable if offering additional findings to infants having sequencing for a clinical indication that has made them critically unwell. In addition, we focused this study on parental choice and did not have sufficient reportable findings to comment on the clinical utility of genomic sequencing in newborns; however, studies looking at utility have already been published.8,40
As the use of exome and genome sequencing increases, there is a critical need to better understand what and how much information should be offered, analyzed, and reported in these tests. Due to significant cultural and geographical differences in the use of this technology it is essential to have diverse population-based evidence to support practice. This study provides the first level of evidence on which to build an ethically sound framework for offering additional findings from genomic sequencing to parents of newborns. It also provides invaluable world-first data showing that genomic newborn screening does not have a negative psychosocial impact on parents.
Change history
11 February 2020
The “Baby Beyond Hearing - Decision Aid” supplementary information file was not uploaded to the Genetics in Medicine website when the paper was published. The omission has now been rectified.
References
Stark Z, Tan TY, Chong B, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18:1090–1096.
Ackerman SL, Koenig BA. Understanding variations in secondary findings reporting practices across U.S. genome sequencing laboratories. AJOB Empir Bioeth. 2018;9:48–57.
Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255.
Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, and Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Pediatrics. 2013;131:620–622.
Borry P, Evers-Kiebooms G, Cornel MC, Clarke A, Dierickx K. Genetic testing in asymptomatic minors: background considerations towards ESHG Recommendations. Eur J Hum Genet. 2009;17:711–719.
Waisbren SE, Back DK, Liu C, et al. Parents are interested in newborn genomic testing during the early postpartum period. Genet Med. 2015;17:501–504.
Goldenberg AJ, Dodson DS, Davis MM, Tarini BA. Parents’ interest in whole-genome sequencing of newborns. Genet Med. 2014;16:78–84.
Bodian DL, Klein E, Iyer RK, et al. Utility of whole-genome sequencing for detection of newborn screening disorders in a population cohort of 1,696 neonates. Genet Med. 2016;18:221–230.
Berg JS, Agrawal PB, Bailey DB, et al. Newborn sequencing in genomic medicine and public health. Pediatrics. 2017;139:e20162252.
Howard HC, Knoppers BM, Cornel MC, Wright Clayton E, Senecal K, Borry P. Whole-genome sequencing in newborn screening? A statement on the continued importance of targeted approaches in newborn screening programmes. Eur J Hum Genet. 2015;23:1593–1600.
Holmes D. Europe plays catch-up on neonatal screening as US skips ahead. Nat Med. 2012;18:1596.
Victorian Clinical Genetics Services. Newborn screening. 2019. https://www.vcgs.org.au/tests/newborn-bloodspot-screening. Accessed 11 September 2019.
Victorian Infant Hearing Screening Program. 2017. https://www.vcgs.org.au/tests/newborn-bloodspot-screening. Accessed 15 July 2017.
Downie L, Halliday JL, Burt RA, et al. A protocol for whole-exome sequencing in newborns with congenital deafness: a prospective population-based cohort. BMJ Paediatr Open. 2017;1:e000119.
Downie L, Halliday JL, Burt RA, et al. Exome sequencing in infants with congenital hearing impairment: a population based cohort study. Eur J Hum Genet. 2019; https://doi.org/10.1038/s41431-019-0553-8. Accessed 12 December 2019.
Halliday JL, Muller C, Charles T, et al. Offering pregnant women different levels of genetic information from prenatal chromosome microarray: a prospective study. Eur J Hum Genet. 2018;26:485–494.
Berg JS, Foreman AK, O’Daniel JM, et al. A semiquantitative metric for evaluating clinical actionability of incidental or secondary findings from genome-scale sequencing. Genet Med. 2016;18:467–475.
Ceyhan-Birsoy O, Machini K, Lebo MS, et al. A curated gene list for reporting results of newborn genomic sequencing. Genet Med. 2017;19:809–818.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Chew LD, Bradley KA, Boyko EJ. Brief questions to identify patients with inadequate health literacy. Fam Med. 2004;36:588–594.
Marteau TM, Bekker H. The development of a six-item short-form of the state scale of the Spielberger State-Trait Anxiety Inventory (STAI). Br J Clin Psychol. 1992;31 (Pt 3):301–306.
O’Connor AM. Validation of a decisional conflict scale. Med Decis Making. 1995;15:25–30.
Brehaut JC, O’Connor AM, Wood TJ, et al. Validation of a decision regret scale. Med Decis Making. 2003;23:281–292.
Carleton RN, Norton MA, Asmundson GJ. Fearing the unknown: a short version of the Intolerance of Uncertainty Scale. J Anxiety Disord. 2007;21:105–117.
Rigante D, Frediani B, Cantarini L. A comprehensive overview of the hereditary periodic fever syndromes. Clin Rev Allergy Immunol. 2018;54:446–453.
Youngblom E, ParianiM. Knowles JW. Familial hypercholesterolemia. In: Adam MP, Ardinger HH, Pagon RA et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993.
Burnett JR, Bell DA, Hooper AJ, Hegele RA. Clinical utility gene card for: familial hypobetalipoproteinaemia (APOB)—update 2014. Eur J Hum Genet. 2015;23:890.
Stark Z, Schofield D, Martyn M, et al. Does genomic sequencing early in the diagnostic trajectory make a difference? A follow-up study of clinical outcomes and cost-effectiveness. Genet Med. 2019;21:173–180.
Petrikin JE, Cakici JA, Clark MM, et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom Med. 2018;3:6.
Joseph G, Chen F, Harris-Wai J, Puck JM, Young C, Koenig BA. Parental views on expanded newborn screening using whole-genome sequencing. Pediatrics. 2016;137:S36–S46.
Waisbren SE, Weipert CM, Walsh RC, Petty CR, Green RC. Psychosocial factors influencing parental interest in genomic sequencing of newborns. Pediatrics. 2016;137:S30–S35.
Genetti CA, Schwartz TS, Robinson JO, et al. Parental interest in genomic sequencing of newborns: enrollment experience from the BabySeq Project. Genet Med. 2019;21:622–630.
Lakes KD, Vaughan E, Lemke A, et al. Maternal perspectives on the return of genetic results: context matters. Am J Med Genet A. 2013;161a:38–47.
LeRoy BS, McCarthy Veach P, Bartels DM. Genetic counseling practice: advanced concepts and skills. Hoboken, NJ: Wiley-Blackwell; 2010.
Holm IA, Agrawal PB, Ceyhan-Birsoy O, et al. The BabySeq project: implementing genomic sequencing in newborns. BMC Pediatr. 2018;18:225.
Lewis MA, Paquin RS, Roche MI, et al. Supporting parental decisions about genomic sequencing for newborn screening: the NC NEXUS decision aid. Pediatrics. 2016;137 (Suppl 1):S16–S23.
Iskrov G, Ivanov S, Wrenn S, Stefanov R. Whole-genome sequencing in newborn screening—attitudes and opinions of Bulgarian pediatricians and geneticists. Front Public Health. 2017;5:308.
Pereira S, Robinson JO, Gutierrez AM, et al. Perceived benefits, risks, and utility of newborn genomic sequencing in the BabySeq Project. Pediatrics. 2019;143 (Suppl 1):S6–S13.
Anderson JA, Meyn MS, Shuman C, et al. Parents perspectives on whole genome sequencing for their children: qualified enthusiasm? J Medical Ethics. 2017;43:535–539.
Ceyhan-Birsoy O, Murry JB, Machini K, et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the BabySeq Project. Am J Hum Genet. 2019;104:76–93.
Acknowledgements
We thank Ivan Maccioca, Gemma Brett, Yael Prawer, Belinda Creighton, Samantha Ayres, Yana Smagarinsky, Ella Wilkins, Jane Wallace, Lisette Curnow, Anaita Kanga-Parabaita, Rigan Titherleigh, Matthew Hunter, Jonathan Berg, Cynthia Powell, and Heidi Rehm. The study was funded by the State Government of Victoria (Department of Health and Human Services) and the ten member organizations of the Melbourne Genomics Health Alliance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Downie, L., Halliday, J., Lewis, S. et al. Exome sequencing in newborns with congenital deafness as a model for genomic newborn screening: the Baby Beyond Hearing project. Genet Med 22, 937–944 (2020). https://doi.org/10.1038/s41436-019-0745-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0745-1
Keywords
This article is cited by
-
Perception of genomic newborn screening among peripartum mothers
European Journal of Human Genetics (2024)
-
Genomic newborn screening for rare diseases
Nature Reviews Genetics (2023)
-
Expanding the Australian Newborn Blood Spot Screening Program using genomic sequencing: do we want it and are we ready?
European Journal of Human Genetics (2023)
