
Abstract
Purpose
We observed four individuals in two unrelated but consanguineous families from Portugal and Brazil affected by early-onset retinal degeneration, sensorineural hearing loss, microcephaly, intellectual disability, and skeletal dysplasia with scoliosis and short stature. The phenotype precisely matched that of an individual of Azorean descent published in 1986 by Liberfarb and coworkers.
Methods
Patients underwent specialized clinical examinations (including ophthalmological, audiological, orthopedic, radiological, and developmental assessment). Exome and targeted sequencing was performed on selected individuals. Minigene constructs were assessed by quantitative polymerase chain reaction (qPCR) and Sanger sequencing.
Results
Affected individuals shared a 3.36-Mb region of autozygosity on chromosome 22q12.2, including a 10-bp deletion (NM_014338.3:c.904-12_904-3delCTATCACCAC), immediately upstream of the last exon of the PISD (phosphatidylserine decarboxylase) gene. Sequencing of PISD from paraffin-embedded tissue from the 1986 case revealed the identical homozygous variant. In HEK293T cells, this variant led to aberrant splicing of PISD transcripts.
Conclusion
We have identified the genetic etiology of the Liberfarb syndrome, affecting brain, eye, ear, bone, and connective tissue. Our work documents the migration of a rare Portuguese founder variant to two continents and highlights the link between phospholipid metabolism and bone formation, sensory defects, and cerebral development, while raising the possibility of therapeutic phospholipid replacement.
Similar content being viewed by others


INTRODUCTION
The enzyme phosphatidylserine decarboxylase (PISD) is responsible for the conversion of phosphatidylserine (PS) to phosphatidylethanolamine (PE), a process that is essential in all living organisms.1 PE is an abundant phospholipid in cellular membranes and is particularly enriched in mitochondrial membranes. PISD is located in the inner mitochondrial membrane of eukaryotic cells2 and it was shown to be essential for the production of PE in situ.3 Experiments in mice have shown that complete inactivation of PISD causes lethality during embryonic development, with no embryos surviving further than embryonic day 12, and histologic images revealed aberrantly shaped and fragmented mitochondria.4 Although this enzyme has been studied extensively in model organisms due to its essential function in eukaryotes, little is known about the role of PISD in relationship to human health and disease.
Here, we report the identification of a genetic and likely hypomorphic variant in PISD, found in homozygosity in five individuals from three families sharing a severe multisystem disorder involving brain, eye, ear, bone, and connective tissue.
MATERIALS AND METHODS
Patients and families
This study was performed according to the tenets of the Declaration of Helsinki, following the signature of written informed consent forms (including the use of images, if applicable) by the patients and their family members and the approval by the institutional review boards of our respective institutions.
Next-generation sequencing
Genomic DNA was extracted from peripheral blood leukocytes according to standard procedures, and then exome libraries (Agilent SureSelectXT Reagent Kit; Agilent Technologies) were sequenced on an Illumina HiSeq 2500 at the Genomic Technologies Facility in Lausanne, Switzerland. Bioinformatic analyses were performed as described previously.5 Briefly, raw reads were mapped to the human reference genome (hg19/GRCh37) using the Novoalign software (V3.08.00, Novocraft Technologies). Next, Picard (version 2.14.0-SNAPSHOT) was used to remove duplicate reads and Genome Analysis Toolkit (GATK) (version 3.8)6 was used to perform base quality score recalibration on both single-nucleotide variants and insertion–deletions. A VCF file with the variants was generated by HaplotypeCaller. Then, DNA variants were filtered based on quality, frequency in ExAC, gnomAD,7 1000 Genomes,8 ESP (NHLBI Exome Variant Server, http://evs.gs.washington.edu/EVS), GME (GME Variome http://igm.ucsd.edu/gme/index.php), and ABraOM,9 and on predicted impact on protein sequence and messenger RNA (mRNA) splicing. Finally, they were annotated according to a specific in-house pipeline.5
Homozygosity mapping and haplotype analysis
Large segments of homozygosity (>1 Mb) were computed from exome sequencing data with a tool developed in-house, AutoMap (unpublished), enabling the detection of shared regions of homozygosity in multiple probands. Then, sequences were visualized to detect shared haplotypes.
Sanger sequencing
To confirm the PISD variant identified by next-generation sequencing (NGS), polymerase chain reaction (PCR) amplification and Sanger sequencing were performed using standard reagents and conditions. The sequences of the primers used in these experiments are 5′-gagtgggactccaaacacatgt-3′ (forward) and 5′-gaatgcaggctcttccgtctat-3′ (reverse).
In silico analysis of the variant
Annotation of chr22:g.32015826_32015835del was verified by VariantValidator10 and its predicted effect on splicing was assessed with MaxEntScan,11 HumanSpliceFinder,12 SplicePort,13 NNSplice,14 and SpliceAI.15
Minigene splicing assay
The genomic sequences of the PISD gene of one patient and a control individual spanning exon 8 – intron 8 – exon 9 and containing the putative splicing variant were amplified by PCR with oligos carrying the recombinant sites attB1 and attB2 (forward 5′-ggggacaagtttgtacaaaaaagcaggctgctccctgatgtcagtgaac-3′ and reverse 5′-ggggaccactttgtacaagaaagctgggtctagagcgagcccagggctt-3′). This minigene was then cloned into a pDEST26 vector (Gateway cloning system, Thermo Scientific). All plasmids were sequenced to verify the correct insertion of mutated and wild-type DNA fragments. The splicing assay was performed by transiently transfecting HEK293T cells with each minigene plasmid using FuGENE HD (Promega). At 24 hours post-transfection, cells were harvested and total RNA was extracted. This was subsequently reverse-transcribed with GoScript (Promega), using oligo-dT probes. The complementary DNAs (cDNAs) of both plasmids were then amplified with sequence-specific primers (5′-gtacaaaaaagcaggctg-3′ and 5′-gtcattgtaggagccctt-3′). PCR products were separated by agarose gel electrophoresis and fragments were analyzed by Sanger sequencing.
Quantitative real-time PCR assay (qPCR)
The expression levels of the wild-type (WT) construct and the one bearing the deletion (del) were assessed using the SYBR green technology (FastStart Universal SYBR Green, Roche) in a QuantStudio 12K Flex real-time PCR system (Applied Biosystems). RPLPO andGAPDH were used as endogenous reference genes to normalize the results. A specific primer pair was used for the amplification of the correctly spliced transcript (forward 5′-ccatcaccatcaccatcactc-3′ and reverse 5′-gccttgggctgtttgtgt-3′). The size of the amplified products was verified by the presence of a single melting peak at the appropriate temperature and their relative expression level was assessed by the ddCt method and by taking into account efficiency of each primer pair. Significance of the difference between two groups was calculated with unpaired Student’s t-test, assuming equal variance.
Targeted variant sequencing of DNA from paraffin-embedded tissue
DNA was extracted from paraffin sections of tiny esophageal biopsies using Nucleospin DNA FFPE (Macherey-Nagel) following manufacturer guidelines. The high-quality DNA was then PCR amplified and sequenced by capillary electrophoresis using the following primers: forward 5′-gcgtcacgaagctgaagtca-3′ and reverse 5′-gagtgggactccaaacacatgt-3′.
Estimation of the degree of consanguinity between affected individuals 1 to 4
In general, an individual having parents who are siblings is expected to have 100/4 = 25% of autozygous regions across their entire genome, and 100/16 = 6.25% if the parents are first cousins. This can be generalized to 100/2n for parents distant by n degrees of relationship (meioses).16 In this report, affected individuals 1, 2, and 3 shared a common autozygous haplotype of 3.36 Mb, corresponding to approximately 0.1% of the human genome. We calculated that this haplotype’s size corresponds to 9.92 estimated meioses, suggesting that the Portuguese and Brazilian patients had a common ancestor approximately five generations ago. Of note, these data do not allow us to estimate the age of the variant itself, which might be significantly older.
RESULTS
Clinical evaluation
Family 1: patients 1 and 2
Patient 1 is a currently 22-year-old man of Portuguese origin, the first child of a healthy couple where parents are second cousins once removed; the maternal grandfather and the paternal great-grandmother were siblings (Fig. 1a). He was referred to genetics at age 2 years 10 months for short stature, generalized joint laxity, and suspicion of skeletal dysplasia. He was born at 38 weeks after an uneventful pregnancy by Cesarean section for pelvic presentation. At birth, weight was 3.4 kg (0 SD), length 47 cm (P5, −1.7 SD), and occipital–frontal circumference (OFC) 37 cm (+1.5 SD). He was admitted to the neonatal unit for 2 days because of respiratory difficulties, with a favorable clinical evolution. Investigation at this point consisted of a karyotype (46,XY), transfontanellar and abdominal ultrasound with normal results, hemogram and thyroid function with normal results, and skeletal survey. Postnatal growth was below −2 SD for length/stature with bilateral hip dislocation as well as severe kyphoscoliosis developing during childhood. Kyphoscoliosis was surgically corrected at age 7 years. Bilateral hip dislocation was treated by traction followed by surgical repositioning with subsequent relapse requiring an additional intervention with osteotomy. Elbow joint dislocation became more pronounced. Height at 11 years was 98.5 cm (P ≪ 3, −7.54 SD); current adult height is 130 cm (P ≪ 3, −7.55 SD). Notably, the cranial circumference growth curve also drifted toward the 5th percentile.

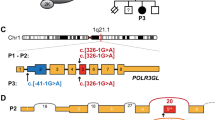
Pedigrees of the three families segregating the Liberfarb syndrome associated with homozygosity for an intronic deletion inPISD. (a) Family 1 is from Portugal; (b) family 2 from Brazil; and (c) family 3, who was the object of the first clinical description of this condition,17 resided in the United States but originated from the Azores islands. (d) Sanger sequencing of the variant, in controls and patients.
Developmental milestones were delayed (head control at 3 months; sitting alone at 8/9 months; first words after 2 years; walking alone at around 4 years), possibly in part secondary to orthopedic findings. Learning difficulties were noted at preschool and a developmental evaluation at 5 years 1 month revealed a global IQ of 50.1; at 7 years 6 months IQ was 68.8 and at the age of 16 years a WISC-III testing revealed a moderate intellectual deficit with a verbal IQ of 46; attention deficit was also reported.
An ophthalmologic evaluation at age 7 revealed best corrected visual acuity (BCVA) of 20/800 for distance and 20/250 for near, with no significant refractive error. A horizontal/torsional manifest nystagmus was detected after age 5 years, which dampened in convergence and in lateroversion. After age 9 years, it became obvious that the proband was mostly using an eccentric point of fixation. Fundus examination disclosed optic disc pallor, generalized mottling of the retinal pigment epithelium (RPE) with areas of atrophy interspersed with pigmentary changes. Bone spicules were identified in the midperiphery and vascular caliber was reduced, rapidly evolving to peripheral avascular areas with remaining ghost vessels (Fig. 2d). These findings, in association with a nonrecordable electroretinogram (ERG) at age 8 years, are compatible with a diagnosis of severe early-onset retinal degeneration (EORD). At age 7 years, suspicion of hypoacusis was substantiated with an audiogram revealing an auditory threshold at 60 dB bilaterally. Subsequently, evoked acoustic potentials confirmed bilateral sensorineural hearing loss with electrophysiological thresholds at 50 and 60 dB respectively in the right and left ears, and hearing aids were prescribed. More recently, symptoms of nasal obstruction have appeared due to hypertrophy of conchae. No other health problems were noted. Neurological examination was unremarkable. Dentition was normal.

Clinical synopsis. (a–c) Patient 4 at the age of 9 years. There is a marked spinal deformity with forward tilting of the pelvis (because of bilateral hip joint dislocation), exaggerated lumbar lordosis, and dorsal kyphosis with scoliosis. Features are identical to those of patient 5 (Fig. 2 in Liberfarb et al., 1986).17 (d) Ocular fundus examination of patient 1 at age 9 years showing optic disc pallor, generalized mottling of the retinal pigment epithelium (RPE) with areas of atrophy interspersed with pigmentary changes, and “bone spicules” identified in the macula and the midperiphery. Vascular caliber was reduced, with peripheral areas being avascular with remaining ghost vessels. (e) Fundus of patient 2 at age 6 years showing atrophy of the optic disc, extremely thin vessels, atrophic central macular area with pigment clumping. (f–k) Skeletal features including bilateral hip dislocation with femoral head dysplasia (patient 3, age 7 years), dislocation of ulna and radial head at the elbow (patient 5, age 15 years), severe epiphyseal dysplasia with striations of metaphyses (patient 3, age 7 years), delay in carpal and phalangeal ossification but no marked dysplasia (patient 3, age 2 years), platyspondyly but no spinal deformity at age 2 months, and extremely severe spinal deformity at adult age (patient 5).
Patient 2 is the younger brother of patient 1. Pregnancy was normal including third trimester ultrasound. He was born at 38 weeks by Cesarean section with weight 3.2 kg (P25), length 47 cm (P < 5, −1.7 SD), OFC 36.5 (P50–75, approximately +1 SD), and Apgar 10/10, and was admitted to the neonatal unit for hypotonia for 7 days with a good evolution. He was referred to genetics at age 4 months because of short stature and short limbs. At age 9 months he had short limbs, short thorax, short neck, and joint laxity with limitation of elbow extension. While global development was normal at 9 months, later language development was delayed. A global development evaluation at 4 years revealed an IQ of 66; at 7 years his IQ was 52, with attention deficit also reported; at 11 years his global IQ was 41. Sensorineural hearing loss was diagnosed at the age of 3 years. Ophthalmological examinations revealed a similar, yet more severe, ocular phenotype as his older sib, since a sensory horizontal nystagmus was noted at age 2 years. At his last examination at age 6 years, he presented a BCVA of 20/400 for distance and 20/200 for near. Fundus examination disclosed findings compatible with severe EORD, revealing bilateral optic atrophy, areas of RPE atrophy and hyperpigmentation in the macula, peripheral bone spicules, and thin retinal vessels (Fig. 2e). At 6 years, seizures with fever occurred and he was admitted to hospital for encephalitis. Orthopedic complications included knee dislocation with valgus left knee, bilateral necrosis of femoral head and pes planus, with surgery performed on left knee and feet, and mild scoliosis. Current weight (age: 12 years) is 27 kg (P < 5; −3 SD), current height is 121 cm (P ≪ 5, −4.32 SD).
Family 2: patients 3 and 4
These two affected brothers were born to healthy Brazilian parents who were first cousins (Fig. 1b). Patient 3 was born at term, after an uneventful pregnancy. At birth, weight was 2.8 kg (P10; −1.5 SD), length 44 cm (P < 3; −2.2 SD), and OFC 36 cm (P75; +1 SD). Motor development was retarded: sitting and walking without support were possible at the ages of 1 year 6 months and 4 years respectively. Scoliosis was noted by the parents “very early.” Clinical examination at the age of 14 years 6 months revealed the following values: height 91.5 cm (P ≪ 2; approximately −8 SD), weight 12.4 kg (P ≪ 2; approximately −9 SD), and OFC 49.5 cm (P ≪ 2; −4 SD). He had severe scoliosis, lumbar lordosis, thoracic kyphosis, bilateral hip dislocation, and bilateral impairment of elbow extension. Language was rudimentary.
Patient 4 was born after an unremarkable full-term pregnancy. Primary adaptation was good. At birth, weight was 3.0 kg (P25; −1 SD), length 44 cm (P < 3; −2.2 SD), and OFC 36 cm (P75). He sat and walked without support at the ages of 1 year 3 months and 3 years, respectively. Scoliosis was observed by the parents since the first year of life. Clinical examination at the age of 11 years 9 months revealed height 100.5 cm (P ≪ 2; approximately −6 SD), weight 15.5 kg (P ≪ 2; approximately −5 SD), and OFC 48 cm (P ≪ 2; approximately −5 SD). He had scoliosis, lumbar lordosis, and bilateral impairment of elbow extension (Fig. 2a–c). There was developmental delay (not formally measured) and very poor language development.
In both patients 3 and 4, brainstem auditory potentials were normal. There was a concern for bilateral cataracts in patient 3, but a formal ophthalmological evaluation could not be performed. However, brain magnetic resonance image (MRI) showed, in both sibs, bilateral optic nerve atrophy and cerebellar atrophy affecting the upper portion of the vermis and the hemispheres. Neurological examination including electromyography (EMG) and neural conduction studies ruled out myopathy and peripheral neuropathy. Patient 4 had onset of seizures at the age of 11 years and has been on anticonvulsant medication since.
Family 3: patient 5
Detailed clinical features of this patient as a girl were published in 1986.17 The patient continued to be followed after initial publication until her death at age 35 years. Briefly, the girl was born to consanguineous parents who had immigrated to the United States from the Azores islands (Portugal). She had first been hospitalized in Portugal at age 17 months for failure to thrive; short stature was noted with marked delay of bone age. Repeat hospitalization at 3 years 8 months of age noted short stature (P < 5), early retinal changes, 25° thoracic scoliosis, lordosis, and hyperextensible joints including marked genu valgum and dislocatable hips. At time of her first hospitalization in Boston (Boston Children’s Hospital), at age 7 years 6 months, examination continued to show short stature as well as respiratory difficulty with subglottic tracheal stenosis, tapetoretinal degeneration, sensorineural hearing loss, and developmental delay. Musculoskeletal problems included thoracic scoliosis increased to 90°, bilateral genu valgum passively positioned to 60°, and bilateral dislocated patellae, dislocated hips, and elbow deformities with dislocated radial heads. Initial surgical management included tracheostomy, bilateral Roux–Goldthwait procedures for patellar dislocation and posterior spinal fusion. In summary, the main observations in this patient were short stature, severe scoliosis, joint laxity, skeletal dysplasia, pigmentary degeneration of the retina, severe sensorineural hearing loss, and moderate developmental delay. No additional orthopedic procedures were performed after spinal fusion as described in the original report. Orthotic support continued over several years with bilateral knee–ankle–foot orthoses and spinal support thoracic–lumbar–sacral orthoses. The patient died at age 35 years following cardiac arrest and anoxic encephalopathy.
The five patients shared a similar clinical pattern. One component was a progressive disease of bone and connective tissue. At birth, there was no scoliosis and no overt joint dislocations. During childhood, the combination of epiphyseal dysplasia and joint laxity resulted in luxation of the femoral head and even more significantly in progressive, severe spinal deformation with lumbar hyperlordosis, thoracic kyphosis, and variable degree of scoliosis (Fig. 2). Joint laxity at the knee joint resulted in severe genua valga. Luxation of the radial head also developed in the first decade. Essential radiographic features were delayed vertebral ossification (but no platyspondyly); markedly delayed ossification of all epiphyses, contributing to joint instability and retarded bone age; and fine metaphyseal striations observed in childhood x-rays, especially at the knees (Fig. 2f–k). The formal classification is that of a spondyloepimetaphyseal dysplasia with predominantly epiphyseal involvement; however, as confirmed by the follow-up of patient 5, severe joint instability with joint dislocations and progressive severe spinal deformation were clinically more significant than the skeletal dysplasia. The second cardinal feature in affected individuals was retinal degeneration. This became clinically apparent in childhood. Detailed ophthalmological evaluation was available for patients 1, 2, and 5 (the findings in patient 5 were published in 1986).17 Patients 3 and 4 had optic atrophy on MRI. The fundus findings were pale optic disks, RPE mottling, severely reduced caliber of the retinal vessels, and areas of bone spicule pigment deposition (Fig. 2d, e). ERG was nonrecordable in patient 1 at age 8 years. These findings are compatible with EORD.
Similarly, microcephaly was not present at birth but was present in all patients at school age, when it also became clear that developmental delay was significant. The data about hypoacusis are unfortunately not very detailed but are compatible with moderate to severe early-onset loss of hearing, rather than with congenital deafness. Thus, the Liberfarb syndrome appears to be a progressive disorder involving connective tissue, bone, retina, ear, and brain.
Genetic and molecular findings
Next-generation sequencing
Because of the association of spondyloepimetaphyseal dysplasia and severe joint laxity, and in spite of the pedigree suggestive of recessive inheritance, individuals 1 to 4 were originally evaluated for the presence of monoallelic KIF22 variants associated with spondyloepimetaphyseal dysplasia with joint dislocations, leptodactylic type (SEMDJL; MIM 603546); no such variants were detected. Subsequently, exome sequencing was performed on three affected children, two from the Portuguese family (patients 1 and 2, Fig. 1a) and one from the Brazilian family (patient 3, Fig. 1b). As parental consanguinity had been reported in both families, we searched for large segments of homozygosity and found two regions that were homozygous in these three subjects (all positions refers to genome build hg19): chr1:66,067,109-84,880,380 (18.8 Mb) and chr22:28,389,453-34,022,284 (5.63 Mb). No rare (allele frequency <1% in available databases and in-house exomes) coding variants were detected in these regions. Through sequence comparison, we found that a part of the region on chromosome 22 was not only fully homozygous in all three individuals, but also shared the same haplotype in the three children (chr22:29,456,733-32,811,952, 3.36 Mb). The investigation for noncoding rare variants identified only one homozygous variant: chr22:g.32015826_32015835del; NM_014338.3:c.904-12_904-3delCTATCACCAC, p.(?). This 10-bp deletion, located in intron 8 of the PISD gene at positions from −3 to −12 before the splice acceptor site of the last exon of the gene (exon 9), was predicted to moderately impair the correct splicing of intron 8 by five different splicing predictors (Supplementary Table 1). Familial analysis confirmed that the variant cosegregated with the disease in both pedigrees, according to a recessive pattern of inheritance (Fig. 1). The exome sequence data was reanalyzed for other putatively pathogenic variants in the shared haplotype region, and none was identified. Targeted Sanger sequencing of the DNA of individual 5 (Fig. 1c), extracted from a paraffin-embedded surgical biopsy of the esophagus, confirmed the presence of the same 10-bp deletion identified in the other affected individuals (Fig. 1d).
Splicing alterations resulting from the variant in PISD
To functionally test the putative consequences of the deletion and in absence of suitable patient-derived material, we performed minigene-based splicing experiments. We designed a minigene plasmid bearing the last two exons of PISD, with and without the microdeletion identified in the patients, for the purpose of expression in a mammalian cell line (Fig. 3a). After plasmid transfection and incubation in HEK293T cells, we examined transcripts originating from plasmids bearing the pathogenic variant versus their wild-type counterpart. We found that the deletion prevented the proper recognition of the natural acceptor splice site of intervening sequence 8 and led to production of both correctly spliced mRNA and transcripts bearing the full retention of intron 8 (Fig. 3b, c, Supplementary Fig. 1). In turn, this latter event resulted in the creation of a premature stop codon within this intron, and possibly to nonsense-mediated decay (NMD)–triggered degradation. If, for any reason, these aberrant transcripts were to escape NMD mechanisms,18 their further translation into a protein is predicted to produce a truncated protein lacking the last 74 amino acids, including its portion containing the decarboxylase alpha chain and the cleavage site for autocatalysis, as predicted by UniProt.19

Schematic view of thePISDgene with the position of the variant identified and the splicing alteration resulting from it. (a) The variant is a 10-bp deletion in intron 8, close to the splice acceptor site of the last exon. The change in the splicing consensus sequence at this site leads to the production of both a canonical transcript and an aberrant messenger RNA (mRNA) retaining intron 8, which in turn leads to a premature termination of the open reading frame. (b) Electrophoresis of the reverse transcription polymerase chain reaction (RT-PCR) products from HEK293T transfected with a minigene-bearing plasmid, carrying exons 8 to 9 of PISD and the 10-bp deletion (del) or the wild-type sequence (WT). The mutant form of the plasmid reveals two transcripts, corresponding to the normally spliced isoform (226 bp) and the one carrying the intronic retention (942 bp) depicted above. (c) Sanger sequencing of the two transcripts, from the WT minigene and the one carrying the deletion, respectively. (d) Quantification of spliced transcripts originating from the WT or mutant plasmid (del), by quantitative PCR (qPCR). The difference between these two values is statistically significant (p-value = 2.5 × 10−4, by t-test). Error bars represent standard deviation values.
Quantitative PCR analyses in this in vitro model indicated that the proportion of correctly spliced mRNA transcripts from plasmids bearing the deletion was only 5.7% compared with transcripts from plasmids carrying the wild-type minigene (Fig. 3d).
DISCUSSION
The four living individuals we studied shared a complex multisystem phenotype including microcephaly, early-onset retinal degeneration, hearing loss, intellectual disability, severe joint laxity, and short stature with skeletal dysplasia. This combination of clinical features is unusual and led us to speculate that they could share a common molecular basis, possibly the same described more than 30 years ago by Liberfarb and coworkers in a single case (individual 5 in this report).17 We also noted that the three families seemed to share a Portuguese ethnic background (the Azores islands are part of Portugal, and Brazil has a strong Portuguese admixture as a former Portuguese colony). DNA sequencing confirmed that all five patients were homozygous for a single variant inPISD, within a shared haplotype, likely due to the presence of a common ancestor for all of them approximately five generations ago. Importantly, no other rare variant was detected within the parts of autozygous regions covered by exome sequencing, including the one on chromosome 1.
The homogeneity of this rare phenotype (which we propose to call the Liberfarb syndrome) in these independently ascertained patients from three different continents, as well as the genetic data obtained, strongly suggest the causal relationship between the observed PISD variant and the clinical phenotype. The transfection experiments confirm the effect of this intronic deletion on mRNA splicing. However, the pathogenesis of this disorder and its pleiotropism remain unexplained. PISD is a fundamental biosynthetic protein that has been studied extensively in model systems, due to its central role in lipid and membrane biology.1,2,3,4,20,21,22 After synthesis, it is transposed to the inner mitochondrial membrane, where it is responsible for the transformation of phosphatidylserine to phosphatidylethanolamine (PE) by decarboxylation, constituting a major source of PE in cells; the other principal PE-biosynthetic pathway being the CDP-ethanolamine pathway (Fig. 4). These two pathways, occurring in different subcellular compartments, are both required for mammalian viability,4,23,24 evoking the existence of distinct pools of PE with dedicated biological function. For instance, PISD is required to maintain mitochondrial PE (mtPE), as PE imported from the CDP-ethanolamine pathway alone cannot sustain adequate mtPE levels.3,25,26 In turn, mtPE determines key biophysical properties of the mitochondrial membrane, through acting as a modulator in mitochondrial fusion and in the biogenesis of mitochondrial membrane proteins.22,27 Complete knockout of PISD function in mice leads to embryonic lethality combined with aberrant morphology of mitochondria, indicating that this protein is probably essential for survival in humans as well. However, heterozygous animals expressing 50% of normal Pisd mRNA had regular mtPE content in tissues and appeared phenotypically normal.4 The presence of a single functional PISD allele seems to be tolerated also in humans, since several presumably healthy carriers of heterozygous loss-of-function variants are reported in the gnomAD database (32 variants with a cumulative frequency of ~0.05%).7 Based on our data from in vitro splicing experiments, it is likely that homozygotes for the variant identified here do not lack PISD function completely, as a small proportion of full-length transcripts could still be detected. Their phenotype may therefore result from a severe but not complete loss of protein activity.

Major phospholipid biosynthetic pathways, genes involved, and subcellular localization of the encoded enzymes.PTDSS1,32PCYT1A,33 andPISD from this study (all in red font) are associated with syndromic skeletal dysplasias. PE is synthesized from two main pathways: the CDP-ethanolamine pathway (Kennedy pathway),34,35 and the PS decarboxylation (PISD) pathway, which takes place in inner mitochondrial membrane.2,36CDP-etn CDP-ethanolamine;CEPT1 choline/ethanolamine phosphotransferase 1; CHKA,B choline kinase; PCYT1A,B, phosphate cytidylyltransferase; CHPT1 choline phosphotransferase 1; ER endoplasmic reticulum; ETNK2 ethanolamine kinase; MAM mitochondria-associated membranes;PC phosphatidylcholine;PCYT2 phosphate cytidylyltransferase 2, ethanolamine; PE phosphatidylethanolamine; PEMT phosphatidylethanolamine N-methytransferase;PISD phosphatidylserine decarboxylase; PS phosphatidylserine; PTDSS1,2 phosphatidylserine synthase.
Two recent studies reported an association between variants inPISD and human genetic disorders. In one report, a homozygous variant causing a NM_014338.3:c.797G>A, p.Cys266Tyr substitution in PISD was found in two individuals from distantly related families, with a skeletal phenotype classified as SEMD, apparently without other clinically significant manifestations.28 We have been able to review the original radiographs (courtesy of our colleague K. Girisha, Manipal, India), and found that the radiographic features are different; the condition observed by Girisha and coworkers includes large (rather than small) epiphyses and does not include joint dislocations, and those findings are at odds with what we have observed in the cases described here. Absence of extraskeletal manifestations (e.g., in eye or development) also distinguishes that condition from the Liberfarb syndrome. A second report associates the PISD variants NM_014338.3:c.830G>A, p.Arg277Gln and c.697+5G>A in compound heterozygosis in two sisters with a phenotype including short stature, midface hypoplasia, infantile cataracts, hypomyelination, ataxia, and intellectual disability.29 The publication does not provide x-ray images; some skeletal changes are mentioned but were considered “not suggestive of a primary skeletal dysplasia.” The global phenotype in those two sisters was considered reminiscent of the CODAS30,31 and EVEN-PLUS5 syndromes (in spite of those two conditions having significant skeletal changes), caused by biallelic variants in the two mitochondrial chaperones LONP1 andHSPA9, and experimental evidence was obtained suggesting an impairment in the function of the mitochondrial membranes. Furthermore, incubation of patients’ fibroblasts with lysophophatidylethanolamine resulted in an improvement of mitochondrial and lysosomal morphology.29
In summary, the available evidence suggests that recessive PISD variants may be responsible for quite divergent clinical phenotypes, possibly related to the severity of the variants detected, ranging from apparently isolated skeletal dysplasia to multisystemic conditions affecting brain, ear, eye, connective tissue, and bone. Such a clinical spectrum and pleiotropism is unusual and goes beyond what is usually seen in bone dysplasia families. But how can this variability and pleiotropism be explained? Zhao and coworkers29 have suggested a possible link between thePISD-associated condition they have observed and the CODAS and EVEN-PLUS phenotypes, supporting the concept of “mitochondrial chaperonopathies.”5 However, we must remark that variants in two other genes involved in the phospholipid synthesis pathway have also been linked to phenotypes combining skeletal dysplasia and sensory disturbances (eye and ear): biallelic PTDSS1 (phosphatidylserine synthase 1) variants determine Lenz–Majewski hyperostotic dwarfism (MIM 151050),32 and biallelic PCYT1A (CTP-phosphocholine cytidylyltransferase) variants are the cause of spondylometaphyseal dysplasia with cone–rod dystrophy (MIM 608940).33 While both enzymes are also expressed in the mitochondrion, the functional relationship between PISD, PCYT1A, and PTDSS1 remains to be clarified. Of note, the precise pathogenesis of either the mitochondrial chaperonopathies or the phospholipid synthesis disorders listed here remains largely unexplained. As a mere hypothesis, we speculate that the PISD protein might have both a metabolic role (in producing PE) and a structural role in the inner mitochondrial membrane, and that different pathogenic variants might have different functional, and thus phenotypic consequences.
In conclusion, in this study we identified the molecular cause of a multiorgan condition, which we suggest calling the Liberfarb syndrome in honor of the ophthalmologist who first described it in 1986. This disorder is caused by a specific variant that appears to have been inherited from an individual of Portuguese origin, and has subsequently spread to three geographical regions. The pathogenesis remains unclear, but the accumulating evidence, including other rare families segregating pathogenic variants in PISD, points to a pleiotropic and variable phenotypic spectrum possibly related to mitochondrial dysfunction (“mitochondrial chaperonopathies”) and to phospholipid synthesis disorders. Much work remains to be done to elucidate molecular pathogenesis and genotype–phenotype correlations in these two groups of disorders. At the end of this road, exogenous replacement with specific phospholipids might be a welcome therapeutic outcome.
Change history
15 January 2020
The original version of the online Article did not contain a space in the title between the words "the" and "PISD". The problem did not occur in the PDF version. This has now been corrected in HTML version of the Article.
References
Lykidis A. Comparative genomics and evolution of eukaryotic phospholipid biosynthesis. Prog Lipid Res. 2007;46:171–199.
Zborowski J, Dygas A, Wojtczak L. Phosphatidylserine decarboxylase is located on the external side of the inner mitochondrial membrane. FEBS Lett. 1983;157:179–182.
Shiao YJ, Lupo G, Vance JE. Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J Biol Chem. 1995;270:11190–11198.
Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem. 2005;280:40032–40040.
Royer-Bertrand B, Castillo-Taucher S, Moreno-Salinas R, et al. Mutations in the heat-shock protein A9 (HSPA9) gene cause the EVEN-PLUS syndrome of congenital malformations and skeletal dysplasia. Sci Rep. 2015;5:17154.
Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 10 11–33.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Naslavsky MS, Yamamoto GL, de Almeida TF, et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat. 2017;38:751–763.
Freeman PJ, Hart RK, Gretton LJ, Brookes AJ, Dalgleish R. VariantValidator: accurate validation, mapping, and formatting of sequence variation descriptions. Hum Mutat. 2018;39:61–68.
Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–394.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67
Dogan RI, Getoor L, Wilbur WJ, Mount SM. SplicePort—an interactive splice-site analysis tool. Nucleic Acids Res. 2007;35:W285–W291.
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–323.
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–548 e524.
Hartl DN, Clark AG. Principles of population genetics. Sunderland, MA: Sinauer Associates; 1997.
Liberfarb RM, Katsumi O, Fleischnick E, Shapiro F, Hirose T. Tapetoretinal degeneration associated with multisystem abnormalities. A case report. Ophthalmic Paediatr Genet. 1986;7:151–158.
Lindeboom RG, Supek F, Lehner B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet. 2016;48:1112–1118.
UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2018;46:2699.
Di Bartolomeo F, Wagner A, Daum G. Cell biology, physiology and enzymology of phosphatidylserine decarboxylase. Biochim Biophys Acta. 2017;1862:25–38.
Percy AK, Moore JF, Carson MA, Waechter CJ. Characterization of brain phosphatidylserine decarboxylase: localization in the mitochondrial inner membrane. Arch Biochem Biophys. 1983;223:484–494.
Chan EY, McQuibban GA. Phosphatidylserine decarboxylase 1 (Psd1) promotes mitochondrial fusion by regulating the biophysical properties of the mitochondrial membrane and alternative topogenesis of mitochondrial genome maintenance protein 1 (Mgm1). J Biol Chem. 2012;287:40131–40139.
Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, Vance JE. Phosphatidylethanolamine deficiency in mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem. 2013;288:4158–4173.
Fullerton MD, Hakimuddin F, Bakovic M. Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2). Mol Cell Biol. 2007;27:3327–3336.
Burgermeister M, Birner-Grunberger R, Nebauer R, Daum G. Contribution of different pathways to the supply of phosphatidylethanolamine and phosphatidylcholine to mitochondrial membranes of the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2004;1686:161–168.
Kainu V, Hermansson M, Hanninen S, Hokynar K, Somerharju P. Import of phosphatidylserine to and export of phosphatidylethanolamine molecular species from mitochondria. Biochim Biophys Acta. 2013;1831:429–437.
Becker T, Horvath SE, Bottinger L, Gebert N, Daum G, Pfanner N. Role of phosphatidylethanolamine in the biogenesis of mitochondrial outer membrane proteins. J Biol Chem. 2013;288:16451–16459.
Girisha KM, von Elsner L, Neethukrishna K, et al. The homozygous variant c.797G>A/p.(Cys266Tyr) in PISD is associated with a spondyloepimetaphyseal dysplasia with large epiphyses and disturbed mitochondrial function. Hum Mutat. 2019;40:299–309.
Zhao T, Goedhart CM, Sam PN, et al. PISD is a mitochondrial disease gene causing skeletal dysplasia, cataracts, and white matter changes. Life Sci Alliance. 2019;2:e201900353
Dikoglu E, Alfaiz A, Gorna M, et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am J Med Genet A. 2015;167:1501–1509.
Strauss KA, Jinks RN, Puffenberger EG, et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+Lon protease. Am J Hum Genet. 2015;96:121–135.
Sousa SB, Jenkins D, Chanudet E, et al. Gain-of-function mutations in the phosphatidylserine synthase 1 (PTDSS1) gene cause Lenz-Majewski syndrome. Nat Genet. 2014;46:70–76.
Hoover-Fong J, Sobreira N, Jurgens J, et al. Mutations in PCYT1A, encoding a key regulator of phosphatidylcholine metabolism, cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94:105–112.
Bakovic M, Fullerton MD, Michel V. Metabolic and molecular aspects of ethanolamine phospholipid biosynthesis: the role of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2). Biochem Cell Biol. 2007;85:283–300.
Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem. 1956;222:193–214.
Borkenhagen L, Kennedy EP, Fielding L. Enzymatic formation and decarboxylation of phosphatidylserine. J Biol Chem. 1961;236:PC28.
Acknowledgements
We thank Brenda J. Barry, Mark D. Fleming, and the Boston Children’s Hospital Department of Pathology for their assistance and contributions. This work was supported by the Swiss National Science Foundation (grant 176097) to C.R.; the Jürg Tschopp MD–PhD Scholarship to V.G.P.; the PhD Fellowship in Life Sciences (Faculty of Biology and Medicine, University of Lausanne) to M.Q.; the Fondation Guillaume Gentil (supporting the Division of Genetic Medicine in Lausanne); the National Institutes of Health (NIH) grant U53HD090255 to Boston Children’s Hospital Department of Neurology; the Japan Agency for Medical Research and Development (AMED) (grants JP18ek0109280, JP18dm0107090, JP18ek0109301, JP18ek0109348, and JP18kk020500 to N. Matsumoto); and the Japan Society for the Promotion of Science (JSPS) KAKENHI (grants JP17H01539 to N. Matsumoto and JP16H05357 to N. Miyake).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Peter, V.G., Quinodoz, M., Pinto-Basto, J. et al. The Liberfarb syndrome, a multisystem disorder affecting eye, ear, bone, and brain development, is caused by a founder pathogenic variant in the PISD gene. Genet Med 21, 2734–2743 (2019). https://doi.org/10.1038/s41436-019-0595-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0595-x
Keywords
This article is cited by
-
Brain imaging findings in Liberfarb syndrome: hypomyelination and optic nerve and cerebellar atrophy
Pediatric Radiology (2022)
-
Biallelic variants in coenzyme Q10 biosynthesis pathway genes cause a retinitis pigmentosa phenotype
npj Genomic Medicine (2022)
-
AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data
Nature Communications (2021)
-
Broadening INPP5E phenotypic spectrum: detection of rare variants in syndromic and non-syndromic IRD
npj Genomic Medicine (2021)
