
Abstract
Purpose
Clinical and experimental evidence has suggested pathobiological crosstalk between lysosomal storage diseases (LSDs) and cancer. We aimed to elucidate the association between germline variants in LSD genes and cancer.
Methods
We performed aggregate rare variant association analysis of potentially pathogenic variants (PPVs) in 42 LSD genes and >30 histological types of cancer using genome sequencing data from 2567 cancer patients (Pan-Cancer cohort) and 2504 healthy individuals (1000 Genomes cohort) and exome sequencing data from 53,105 individuals without cancer (ExAC cohort).
Results
PPVs were significantly enriched in the Pan-Cancer cohort compared with the 1000 Genomes cohort (PPV prevalence, 20.7% vs. 13.5%; P = 8.7 × 10−12). Cancer risk was higher in individuals with a greater number of PPVs (P = 7.3 × 10−12). Population structure–adjusted optimal sequence kernel association test (SKAT-O) revealed 37 significantly associated cancer type–LSD gene pairs. These results were supported by the consistent tendency toward enrichment of PPVs in cancer patients compared with the ExAC cohort. Cancer developed earlier in PPV carriers than in wild-type patients. Analysis of tumor transcriptomic data from the pancreatic adenocarcinoma cohort revealed 508 genes differentially expressed according to PPV carrier status, which were highly enriched in the core signaling pathways of pancreatic cancer.
Conclusion
Carriers of PPVs in LSD genes are at increased risk of cancer.
Similar content being viewed by others

INTRODUCTION
Lysosomal storage diseases (LSDs) comprise more than 50 disorders caused by inborn errors of metabolism, which involve the impaired function of endosome–lysosome proteins.1 In LSDs, defects in genes encoding lysosomal hydrolases, transporters, and enzymatic activators result in macromolecule accumulation in the late endocytic system.2 The disruption of lysosomal homeostasis is linked to the increased endoplasmic reticulum and oxidative stress,3 proliferative signaling,4 degradation of the extracellular matrix,5,6 lysosomal exocytosis,7 and integrin β4-mediated cellular migration and invasion,8 all of which can induce oncogenic cellular phenotype and promote the development of malignancy.9
Typical LSD patients have severely impaired organ functions and short life expectancy. However, a considerable number of undiagnosed LSD patients have mildly impaired lysosomal function and survive into adulthood.1 These patients are often diagnosed after they develop secondary diseases such as Parkinsonism that are attributable to insidious LSDs.10 Clinical observations have shown that patients with Gaucher disease or Fabry disease are at increased risk of cancer,11,12 indicating that dysregulated lysosomal metabolism may contribute to carcinogenesis. However, the precise relationship between lysosomal dysfunction and cancer remains unclear; this uncertainty can be attributed in part to the diverse and nonspecific phenotypes of LSDs and the resulting difficulty in recognizing patients with mild symptoms. The extensive allelic heterogeneity and the complex genotype–phenotype relationships make the diagnosis more challenging.13 Furthermore, growing evidence suggests that single allelic loss is functionally significant, though the impact may not be sufficient to develop the overt disease.14 Considering the above along with the recessive inheritance nature of most LSDs, we hypothesized that there would be a large number of undetected carriers of causal variants of LSDs with mild functional impairment, and these carriers would be at increased risk of cancer.
Here we report the results of a comprehensive association analysis between cancer and germline variants in causal genes of LSDs using data from international sequencing projects. We show that carriers of potentially pathogenic variants (PPVs) in LSD genes are at increased risk of cancer, cancer risk is higher in individuals with a greater number of PPVs, cancer develops earlier in PPV carriers, and the transcriptional misregulation of cancer-promoting signaling pathways might underlie the oncogenic contribution of PPVs. Potential carcinogenic mechanisms were investigated using tumor genomic and transcriptomic data with a focus on the pancreatic adenocarcinoma.
MATERIALS AND METHODS
Study populations
We used matched tumor-normal pair genome and tumor transcriptome sequencing and clinical data of 2567 cancer patients (Pan-Cancer cohort) from the Pan-Cancer Analysis of Whole Genomes (PCAWG) project (P.J. Campbell et al., unpublished data). As controls, we used publicly available variant call sets from two global sequencing projects of individuals without known cancer histories. The first control data set comprised 2504 genomes from the 1000 Genomes Project (1000 Genomes cohort).15 The second data set was derived from exomes of 53,105 unrelated individuals from a subset of the Exome Aggregation Consortium release 1.0 that did not include the Cancer Genome Atlas (TCGA) subset (ExAC cohort).16
Potentially pathogenic variant selection
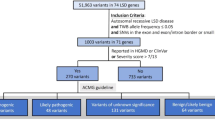
We identified 42 genes implicated in the development of LSDs via comprehensive literature review (Table 1).1,13,17,18,19 Within the genomic loci of those genes, we selected PPVs using three different measures to determine their pathogenicity: (1) predicted consequences on transcripts or protein products they encode (tier 1); (2) clinical and experimental evidence obtained from curated variant databases including ClinVar, Human Gene Mutation Database, and locus-specific mutation databases (tier 2; Supplementary Table S1);20,21 and (3) in silico prediction of variant effects on protein function (tier 3; Supplementary Figure S1). An average allele frequency cutoff of 0.5% (between the case and control cohorts) was also used to filter out common (hence likely nonpathogenic) variants (Fig. 1a).

Potentially pathogenic variant (PPV) selection criteria and population composition of the Pan-Cancer and 1000 Genomes cohorts. (a) Venn diagram of PPVs identified in the Pan-Cancer and 1000 Genomes cohorts grouped into three tiers. (b,c) Populations comprising the Pan-Cancer cohort (b) and the 1000 Genomes cohort (c). (d) Populations comprising each cancer type of the Pan-Cancer cohort. AFR African, AMR American, ASN East Asian, Biliary-AdenoCA gallbladder and extrahepatic bile ducts cholangiocarcinoma, Bladder-TCC urinary bladder transitional cell carcinoma, Bone-Osteosarc bones and joints osteosarcoma, Breast-AdenoCA breast invasive ductal carcinoma, Cervix-SCC uterine cervix squamous cell carcinoma, CNS-GBM central nervous system glioblastoma, CNS-Medullo central nervous system medulloblastoma, CNS-Oligo central nervous system oligodendroglioma, CNS-PiloAstro central nervous system pilocytic astrocytoma, ColoRect-AdenoCA colon and rectum adenocarcinoma, Eso-AdenoCA esophagus adenocarcinoma, EUR European, Head-SCC head and neck squamous cell carcinoma, HGMD Human Gene Mutation Database, Kidney-ChRCC renal cell carcinoma—chromophobe type, Kidney-RCC renal cell carcinoma—clear cell type, Liver-HCC hepatocellular carcinoma, Lung-AdenoCA lung adenocarcinoma, Lung-SCC lung squamous cell carcinoma, Lymph-BNHL non-Hodgkin B-cell lymphoma, Lymph-CLL chronic lymphocytic leukemia, mRNA messenger RNA, Myeloid-AML acute myeloid leukemia, Myeloid-MPN myeloproliferative neoplasm, Ovary-AdenoCA ovary adenocarcinoma, Panc-AdenoCA pancreas adenocarcinoma, Panc-Endocrine pancreas neuroendocrine carcinoma, Prost-AdenoCA prostate adenocarcinoma, SAN South Asian, Skin-Melanoma skin malignant melanoma, SoftTissue-Leiomyo soft tissue leiomyosarcoma, SoftTissue-Liposarc soft tissue liposarcoma, Stomach-AdenoCA stomach adenocarcinoma, Thy-AdenoCA thyroid adenocarcinoma, Uterus-AdenoCA uterus adenocarcinoma, UTR untranslated region.
Statistical analysis
In the primary analysis, the Pan-Cancer and 1000 Genomes cohorts were analyzed by using the optimal sequence kernel association test (SKAT-O) method for aggregate rare variant association of PPVs with cancer,22 and Fisher’s exact tests and logistic regressions for direct comparison of PPV prevalence. We performed Fisher’s exact tests to validate the association detected by the primary analysis using the ExAC cohort as an independent control. The age at diagnosis of cancer was compared between groups using Wilcoxon rank sum tests and linear regression. To improve the statistical power in the analysis of patient age at diagnosis, we combined multiple histological cohorts sharing similar clinicopathologic characteristics into a single clinical cohort (Supplementary Table S2). We assumed that all study individuals represent independent observations as the original study designs did not involve any matching between individuals (P.J. Campbell et al., unpublished data).15,16 Correction for multiple testing was conducted using the false discovery rate (FDR) estimation procedure.23 All tests were two-tailed unless otherwise specified. We considered FDR < 0.1 and P < 0.05 (when not adjusted for multiple testing) significant. Statistical analysis was performed using R version 3.5.0 (R Foundation for Statistical Computing, Vienna, Austria) and Bioconductor version 3.7. For full analytical details, see the Supplementary Materials and Methods.
Ethical approval
Protocols for patient enrollment, sample collection, and data sharing for the PCAWG project were approved by local ethics committees in accordance with the local policies and the bioethical framework of the International Cancer Genome Consortium.24
RESULTS
Characteristics of study cohorts
The Pan-Cancer cohort comprised four populations and 38 histological types of pediatric or adult cancer (Fig. 1b, d and Supplementary Table S2). The median age at diagnosis was 60 years (range 1–90). A majority of the patients were Europeans or Americans in most cancer types. The 1000 Genomes cohort comprised five populations (Fig. 1c);15 we combined the European and American populations for comparison with the Pan-Cancer cohort. The ExAC cohort included seven populations, among which the Americans and non-Finnish Europeans together accounted for more than 60% of the entire cohort.16
PPV prevalence in the Pan-Cancer and 1000 Genomes cohorts
Within the genomic loci of 42 LSD genes (Table 1), 7187 germline single-nucleotide variants and indels were identified in protein-coding regions, essential splice junctions, and 5′ and 3′ untranslated regions in the merged variant call set of the Pan-Cancer and 1000 Genomes cohorts (Supplementary Figure S2). Via an automated algorithm-based approach, a total of 432 PPVs were selected in 41 genes (Fig. 1a); no PPV was identified in LAMP2 (Supplementary Figure S3A and Supplementary Table S3). Overall, the prevalence of PPVs (proportion of individuals carrying PPVs) was significantly higher in the Pan-Cancer cohort than in the 1000 Genomes cohort (20.7% vs. 13.5%; odds ratio, 1.67; 95% confidence interval, 1.44–1.94; P = 8.7 × 10−12; Fig. 2a). This association remained significant after adjustment for population structure (odds ratio, 1.44; 95% confidence interval, 1.22–1.71; P = 2.4 × 10−5). The odds ratio for cancer risk was higher in individuals with a greater number of PPVs (Cochran–Armitage trend test P = 7.3 × 10−12). The results were broadly consistent when the analysis was restricted to individual tiers, although some tier-specific results did not reach statistical significance (Fig. 2a). In contrast, the rare synonymous variants (RSVs) were almost equally prevalent in both cohorts after population structure adjustment (Fig. 2b), demonstrating that (1) the enrichment of PPVs in the Pan-Cancer cohort was not likely due to batch effects, and (2) our strategy to adjust for population structure was working efficiently in the context of rare variants such as PPVs and RSVs. The gene-specific prevalence of PPVs and RSVs in the Pan-Cancer and 1000 Genomes cohorts is shown in Supplementary Figure S3B and C, respectively. The results demonstrated that PPVs were relatively more abundant in the Pan-Cancer cohort versus the 1000 Genomes cohort with respect to the abundance of RSVs, for 33 of 42 genes (78.6%; exact binomial test P < 0.001).

Enrichment of potentially pathogenic variants (PPVs) in cancer patients. (a) Odds ratios for the prevalence of total PPVs (with or without population adjustment) or PPVs belonging to each tier in the Pan-Cancer versus 1000 Genomes cohorts. Odds ratios for the prevalence of single, double, and triple PPV carriers (individuals carrying one, two, or three PPVs, respectively) are also presented without population adjustment. Odds ratios for double and triple carriers of tier 3 PPVs and triple carriers of total PPVs are 7.54, infinite, and 7.4, respectively, with the corresponding bars cut off at the top edge of the plot. (b) Odds ratios for the prevalence of rare synonymous variants (RSVs) analyzed in the same manner as for PPVs. Error bars indicate 95% confidence intervals. (c) Optimal sequence kernel association test (SKAT-O) association between 30 major histological types of cancer (>15 patients per type) and PPVs in each lysosomal storage disease (LSD) gene. Significantly associated cohort–gene pairs at the 0.1 false discovery rate (FDR) threshold are encircled by bold rings. Cohorts are shown in descending order according to the number of patients they include (top to bottom), and genes are shown in descending order according to the number of unique PPVs they contain (left to right). (d) Quantile–quantile plot of P values derived from SKAT-O analyses. A group-based inflation factor (λ) is displayed at the top left-hand corner. Gray shading indicates the 95% confidence interval. Each dot in this plot corresponds to each dot shown in (c). (e) Odds ratios and corresponding 95% confidence intervals for the prevalence of PPVs in eight significant cancer cohorts versus the ExAC control cohort. The right side of the forest plot shows the PPV allele frequency in each cancer cohort and the ExAC cohort. (f) Odds ratios and corresponding 95% confidence intervals for the prevalence of ten significant PPV groups identified in the SKAT-O analysis in the Pan-Cancer versus ExAC cohorts. The right side of the forest plot shows the allele frequency of PPVs belonging to each group in the Pan-Cancer and ExAC cohorts. The gray dashed lines in (e) and (f) represent the odds ratios for the Pan-Cancer cohort and the overall PPV set, respectively. Because there were no tier 3 variants in the validation analysis (see Supplementary Materials and Methods), the PPV frequency in the Pan-Cancer cohort is lower here (8.5%) than in the primary analysis (12.2%). Biliary-AdenoCA gallbladder and extrahepatic bile ducts cholangiocarcinoma, Bladder-TCC urinary bladder transitional cell carcinoma, Bone-Osteosarc osteosarcoma, Breast-AdenoCA breast invasive ductal carcinoma, Cervix-SCC uterine cervix squamous cell carcinoma, CNS-GBM glioblastoma, CNS-Medullo medulloblastoma, CNS-Oligo oligodendroglioma, CNS-PiloAstro pilocytic astrocytoma, ColoRect-AdenoCA colon and rectum adenocarcinoma, Eso-AdenoCA esophagus adenocarcinoma, Head-SCC head and neck squamous cell carcinoma, Kidney-ChRCC renal cell carcinoma—chromophobe type, Kidney-RCC renal cell carcinoma—clear cell type, Liver-HCC hepatocellular carcinoma, Lung-AdenoCA lung adenocarcinoma, Lung-SCC lung squamous cell carcinoma, Lymph-BNHL non-Hodgkin B-cell lymphoma, Lymph-CLL chronic lymphocytic leukemia, Myeloid-AML acute myeloid leukemia, Myeloid-MPN myeloproliferative neoplasm, Ovary-AdenoCA ovary adenocarcinoma, Panc-AdenoCA pancreas adenocarcinoma, Panc-Endocrine pancreas neuroendocrine carcinoma, Prost-AdenoCA prostate adenocarcinoma, Skin-Melanoma skin malignant melanoma, SoftTissue-Leiomyo soft tissue leiomyosarcoma, SoftTissue-Liposarc soft tissue liposarcoma, Stomach-AdenoCA stomach adenocarcinoma, Thy-AdenoCA thyroid adenocarcinoma, Uterus-AdenoCA uterus adenocarcinoma.
Association of PPVs with individual cancer types
Among the 30 major histological types of cancer (>15 individuals per cancer type), the PPV prevalence ranged from 8.8% to 48.6%, with significantly higher values in seven histological types than in the 1000 Genomes cohort (Supplementary Figure S4A). Results of tier-specific analyses were broadly consistent (Supplementary Figures S4B–D). In contrast, RSV prevalence showed much less variation across cohorts and was higher in the 1000 Genomes cohort than in any cancer cohort (Supplementary Figure S4E), reflecting the more heterogeneous nature of the ancestry in the 1000 Genomes cohort resulting in higher genetic diversity (Fig. 1c). SKAT-O adjusted for population structure revealed 37 significantly associated cancer–gene pairs and four genes (GBA, SGSH, HEXA, and CLN3) with a pan-cancer association (Fig. 2c, Supplementary Figure S3B, and Supplementary Table S4). Among those, 36 cancer type–specific associations and 3 pan-cancer associations exhibited a ratio of PPV prevalence in the cancer cohort and the 1000 Genomes cohort exceeding 2 (Supplementary Table S4), suggesting that the average effect size of PPVs for these significant cancer–gene pairs is larger than that of common risk variants identified through the genome-wide association studies.25,26 Overall, 19 cancer types were significantly enriched for PPVs in at least 1 LSD gene, and PPVs in 18 genes were associated with at least one cancer type. We observed no evidence of systematic inflation of test statistics from the SKAT-O results (Fig. 2d).
PPV prevalence in the Pan-Cancer and ExAC cohorts
For the purpose of validation, we focused on (1) eight cancer cohorts that showed significant PPV enrichment compared with the 1000 Genomes cohort (Supplementary Figure S4A) and (2) ten PPV groups that were significantly enriched in the Pan-Cancer cohort or three or more histological cancer subgroups compared with the 1000 Genomes cohort (Supplementary Figure S3B and Fig. 2c). As shown in Fig. 2e, PPV prevalence was higher in all tested cancer cohorts than in the ExAC cohort, and the association was significant for the Pan-Cancer, pancreatic adenocarcinoma, medulloblastoma, pancreatic neuroendocrine carcinoma, and osteosarcoma cohorts. In addition, all tested PPV groups except GBA were more prevalent in the Pan-Cancer cohort than in the ExAC cohort, and six were significantly enriched in cancer patients (Fig. 2f). Although these analyses were not adjusted for population structure due to the lack of individual-level genotype data for the ExAC cohort, the ExAC cohort exhibited a more similar population composition to the Pan-Cancer cohort as compared with the 1000 Genomes cohort (Fig. 1b and Supplementary Figure S5), supporting the relevance of results from the unadjusted analysis.
Subgroup analysis after excluding the ovarian adenocarcinoma and acute myeloid leukemia samples
Recently, Buckley et al. demonstrated that the whole-genome amplification of DNA prior to sequencing could result in serious technical artifacts in the final variant calls using TCGA data set, which might confound the association analysis relating germline variants to tumor types.27 They showed that these artifacts primarily affected the ovarian adenocarcinoma and acute myeloid leukemia samples.27 As almost one-third of the PCAWG germline samples are from TCGA (P.J. Campbell et al., unpublished data), we replicated our pan-cancer analysis after excluding all ovarian adenocarcinoma and acute myeloid leukemia samples from the Pan-Cancer cohort. The results were highly consistent with those of the pan-cancer analysis described above, demonstrating the robustness of the pan-cancer association with PPVs observed in our study (Supplementary Figures S6–8).
Age at diagnosis of cancer according to PPV carrier status
The age at diagnosis of cancer across 28 major clinical cancer cohorts (corresponding to 30 major histological types that included 15 or more patients; information on age at diagnosis was not available for the osteosarcoma cohort) is shown in Fig. 3a. To examine whether cancer occurred earlier in PPV carriers than in wild-type individuals, we first compared the age at diagnosis of cancer according to PPV carrier status in the Pan-Cancer cohort and in six clinical cancer subgroups that showed significant PPV enrichment in the SKAT-O analysis (Fig. 3b). The median age at diagnosis of cancer was numerically lower in PPV carriers in all evaluated cohorts, and the difference was significant in the following cohorts: Pan-Cancer (median age, 59 vs. 61 years; P = 0.002), pancreatic adenocarcinoma (median age, 61 vs. 68.5 years; P < 0.001), and chronic myeloid disorder (median age, 45.5 vs. 58.5 years; P = 0.044). We next compared the age at diagnosis of cancer between carriers and noncarriers of PPVs that belonged to each PPV group that was significantly enriched in the Pan-Cancer cohort or three or more cancer types compared with the 1000 Genomes cohort (Fig. 3c). Among the Pan-Cancer cohort, carriers of PPVs that belonged to tier 1, tier 3, HGSNAT, CLN3, and NPC2 had a significantly earlier diagnosis of cancer compared with wild-type individuals. Moreover, the PPV load consistently showed a negative linear correlation with age at diagnosis of cancer across all cancer types and PPV groups evaluated, and the correlation was significant in the Pan-Cancer and pancreatic adenocarcinoma cohorts (Fig. 3d, e). Exploratory analysis across all cancer types and LSD genes revealed earlier cancer diagnosis in PPV carriers for five additional cancer–gene pairs (Fig. 3f), three of which (pancreatic adenocarcinoma–MAN2B1, cutaneous melanoma–NPC2, and chronic myeloid disorder–SGSH) were in concordance with the SKAT-O results (Fig. 2c).

Age at diagnosis of cancer. (a) Age at diagnosis of cancer across 28 major clinical cancer cohorts. Patients are represented by red (potentially pathogenic variant [PPV] carrier) or gray (noncarrier) dots. Boxes encompass the 25th through 75th percentiles, the horizontal bar represents the median, and the upper and lower whiskers extend from the upper and lower hinges to the largest and smallest values no further than 1.5× interquartile range from the hinges, respectively. Data beyond the end of whiskers are plotted individually. (b) Age at diagnosis of cancer in carriers and noncarriers of PPVs in the Pan-Cancer cohort and six clinical cancer subgroups that showed significant optimal sequence kernel association test (SKAT-O) association with PPVs. (c) Age at diagnosis of cancer according to the carrier status of 11 PPV groups significantly associated with the Pan-Cancer cohort or more than two histological cancer subgroups in the SKAT-O analysis. (d,e) Linear correlations between the PPV load and age at diagnosis of cancer in six clinical cancer subgroups shown in (b) (d) and in the Pan-Cancer cohort for each of 11 PPV groups shown in (c) (e). In (d) and (e), each dot represents a single patient. Simple linear regression was performed for each cohort in (d), and linear regression adjusted for cancer histology was performed for each group of PPVs in (e) to draw the regression line and test for statistical significance. As plots in (d) and (e) are magnified to clearly distinguish between regression lines, not all patient dots are included within the plotted area. (f) All cancer–gene pairs in which age at diagnosis of cancer differs significantly according to the PPV carrier status. In (b), (c), and (f), P values derived from one-sided Wilcoxon rank sum tests are shown above each violin plot. The vertically aligned P values from top to bottom for PACA in (f) correspond to the three genes displayed from left to right, respectively. The red dot in each violin plot represents the median. BLCA bladder urothelial carcinoma, BNHL non-Hodgkin B-cell lymphoma, BRCA breast cancer, BTCA biliary tract cancer, CESC cervical squamous cell carcinoma, CLLE chronic lymphocytic leukemia, CMBT medulloblastoma, CMDI chronic myeloid disorders, COAD colorectal adenocarcinoma, ESAD esophageal adenocarcinoma, GBM glioblastoma multiforme, HNSC head and neck squamous cell carcinoma, KICH chromophobe renal cell carcinoma, KIRC clear cell renal cell carcinoma, LAML acute myeloid leukemia, LGG low-grade glioma, LIHC hepatocellular carcinoma, LUAD lung adenocarcinoma, LUSC lung squamous cell carcinoma, OV ovarian cancer, PACA pancreatic adenocarcinoma, PAEN pancreatic neuroendocrine carcinoma, PRAD prostate adenocarcinoma, SARC soft tissue sarcoma, SKCM cutaneous melanoma, STAD stomach adenocarcinoma, THCA thyroid cancer, UCEC ueterine corpus endometrial carcinoma.
Differential patterns of somatic variants and gene expression in pancreatic adenocarcinomas from PPV carriers versus noncarriers
We sought to determine whether differentiating patterns of somatic variants and gene expression underlie the oncogenic process triggered by PPVs in pancreatic adenocarcinoma, for which both the SKAT-O analysis and comparison of age at diagnosis of cancer according to PPV carrier status produced consistent results (Figs. 2, 3 and Supplementary Figure S4A–D). We first compared the somatic mutational landscape between tumors from PPV carriers (n = 55) versus noncarriers (n = 177). The five most frequently mutated genes were common in both groups (Supplementary Figure S9), and the first four of these were in agreement with the results of previous genome sequencing studies of pancreatic adenocarcinoma.28,29 Nonsilent mutation burden was similar between groups (mean 57.1 vs. 56.3 variants per tumor for PPV-associated vs. PPV-unrelated cases, respectively; P = 0.9). The mutational signature also did not differ by the PPV carrier status (P ≥ 0.05 for all signatures; Supplementary Figure S10).
A differentially expressed gene (DEG) analysis of pancreatic adenocarcinoma samples with available RNA-Seq data (n = 55; 8 from carriers and 47 from noncarriers of PPVs) revealed 287 gene upregulations and 221 downregulations in tumors from PPV carriers versus noncarriers (Fig. 4a–d and Supplementary Table S5). The pathway-based analysis identified 63 pathways significantly altered in PPV carriers (Fig. 4e and Supplementary Figure S11). Remarkably, these pathways included at least 6 among 13 core signaling pathways that had been shown to be recurrently perturbed in pancreatic cancer: Ras signaling, Wnt signaling, axon guidance, cell cycle regulation, focal adhesion, cell adhesion, and extracellular matrix–receptor interaction pathways.29,30 In addition, our data suggested that deleterious variants in LSD genes can provoke perturbations in pathways involved in the pathophysiology of Parkinson disease, Alzheimer disease, and Huntington disease, all of which have been reported to occur frequently in LSD patients.1 The glycerophospholipid metabolism pathway was also identified, indicating the lysosomal dysfunction in PPV carriers.

Differentially expressed genes and pathways in pancreatic adenocarcinoma from potentially pathogenic variant (PPV) carriers versus noncarriers. (a–c) Differentially expressed gene (DEG) analysis reveals 287 gene upregulations and 221 downregulations in PPV-associated pancreatic adenocarcinoma. In (a) and (b), genes with false discovery rate (FDR) <0.1 are shown in red. In (c) the histogram of P values shows a peak frequency below 0.05, demonstrating the existence of up- or downregulated genes. (d) Heatmap showing the relative expression of genes significantly up- or downregulated at the 0.1 FDR threshold in tumors from PPV carriers versus noncarriers, labeled with red and gray bars under the dendrogram, respectively. We ranked the samples according to the FPKM-UQ–normalized read counts for each gene and used the rank numbers for color mapping to standardize the visual contrast across genes. Samples are ordered as columns by hierarchical clustering based on the Euclidean distance and complete linkage. Genes are ordered as rows in the same manner (dendrogram not shown). High and low relative expression is indicated by progressively more saturated red and blue colors, respectively. (e) KEGG pathways that are significantly altered in tumors from PPV carriers compared with those from noncarriers. Only pathways of particular interest discussed in the text are shown. All pathways with FDR <0.1 are shown in the Supplementary Figure S11. ECM extracellular matrix, FC fold change.
DISCUSSION
This study describes the results of comprehensive association analysis of PPVs in LSD genes and cancer across >30 histological types. Our aggregate rare variant association analysis enabled the detection of rare variant enrichment both in the Pan-Cancer cohort and in specific cancer types compared with the cancer-free population, revealing at least twofold enrichment of PPVs in cancer patients for the majority of significantly associated cancer–gene pairs in the SKAT-O analysis. Given the rarity of individual PPVs and the relatively small number of patients included in specific cancer type cohorts, most of these associations would have been left undetected with individual variant-level analyses and were discoverable only because of recent explosive accumulation of genome and exome sequencing data. Findings from the primary analysis were corroborated by (1) consistent results from tier-specific analyses, (2) validation with an independent control cohort, and (3) comparison of the results with those obtained from synonymous variants with matched allele frequency to PPVs. The genetic association was further supported by the significant difference in age at diagnosis of cancer observed in carriers versus noncarriers of PPVs.
Against the traditional view that the biallelic inactivation of tumor suppressor genes is a prerequisite for the malignant cellular transformation, recent research has proposed a continuum model of tumor suppression, emphasizing the crucial impact of a subtle expression change of tumor suppressor genes.31 In line with the rarity of individual PPVs, almost all PPV carriers observed in our study were heterozygous for PPVs in specific genes. Therefore, the dosage effect model may also be useful in explaining the mechanisms involved in the oncogenic contribution of LSD gene variants, as already implied by a previous study.14
From the SKAT-O analysis, we identified four genes that showed a significant pan-cancer association; among those, SGSH and CLN3 were strongly associated with five and four cancer types, respectively. SGSH encodes sulfamidase, a lysosomal hydrolase that degrades heparan sulfate. Deficiency of sulfamidase leads to Sanfilippo syndrome A (mucopolysaccharidosis IIIA), which is characterized by progressive mental and behavioral deterioration that typically presents in childhood. However, an adult-onset disease that presents primarily with visceral manifestations without neurological abnormality has also been reported.32 A recent in vivo study suggested a crucial role of oxidative stress in the pathobiology of Sanfilippo syndrome A.33 Since the oxidative stress is a key mediator of cancer cell growth, invasiveness, and angiogenesis,9 inherited SGSH variants may contribute to an elevated cancer risk via persistent cellular exposure to oxidative stress, a plausible hypothesis that should be confirmed in future functional studies.
CLN3 is a late endosomal and lysosomal transmembrane protein, and its defect causes classic juvenile neuronal ceroid lipofuscinosis (CLN3 disease). In CLN3 disease, impaired trafficking of galactosylceramide to the plasma membrane promotes the generation of proapoptotic ceramide and subsequent activation of caspases, which in turn accelerates apoptosis.18 In line with its control over apoptosis, CLN3 also regulates cancer cell growth, and its therapeutic implication has been suggested.34 Therefore, the results of our study warrant investigation of this protein as a therapeutic target for the treatment of various types of cancer.
Almost 5–10% of pancreatic cancer patients are diagnosed before the age of 50.35 For these patients, positive family history is a strong risk factor, indicating the presence of inherited risk variants.36 Indeed, many risk alleles have been identified in genes involved in the genome maintenance and double-strand DNA break repair (e.g., BRCA1/2 and PALB2). However, in a majority of the early-onset pancreatic cancer patients, the genetic cause remains unclear.37 In our histology-specific analysis, patients with pancreatic adenocarcinoma showed a strong association with PPVs in several LSD genes and had a significantly earlier diagnosis of cancer, motivating us to evaluate differential patterns of somatic variants and gene expression in this histological subset. The DEG analysis revealed many genes up- or downregulated in PPV carriers, and the gene set analysis provided novel insights into the biological processes that might be involved in pancreatic carcinogenesis in these patients. Remarkably, many of the altered pathways identified in the gene set analysis were previously implicated in pancreatic cancer development in transcriptome and exome sequencing studies.29,30 The somatic variants profiles, in contrast, were comparable between the carriers and noncarriers of PPVs. Overall, the results of our study suggest that the transcriptional misregulation acts as a key mediator of pancreatic carcinogenesis triggered by PPVs.
This study has limitations. First, as we did not process the raw sequence data but used variant call sets produced by independent research consortia, the possibility of batch effects exists, even considering the similarity in platforms and pipelines used to generate each data set and the contrasting results from analyses using PPVs versus RSVs (P.J. Campbell et al., unpublished data).15,16 Second, although the ExAC cohort served as a large-scale validation control, we could not perform the SKAT-O analysis with this cohort as control, nor could we adjust for population structure using this cohort because the individual-level genotype data were not accessible. Third, an independent cancer cohort that is sufficiently powered for analyzing such rare variants as PPVs was not available for external validation. Finally, hematological malignancies such as myeloma, the most widely known LSD-associated cancer, were poorly represented in the Pan-Cancer cohort, and the cohort size of specific cancer types was not sufficiently large to draw reliable histology-specific conclusions.
From a therapeutic perspective, LSD genes are attractive targets given the mechanistically intuitive nature of the enzyme replacement and substrate reduction therapies.38 Other promising approaches include pharmacological chaperones, gene therapy, and compounds that “read through” the early stop codon introduced by nonsense variants.38 Although it is unclear whether preemptive treatment can prevent or delay long-term complications of LSDs such as cancer, our findings make it promising to harness these sophisticated LSD therapies for preventing cancer in carriers of inactivating LSD gene variants.
In conclusion, the present study provides a comprehensive landscape of association between PPVs in LSD genes and cancer. Investigating the crosstalk between treatable metabolic diseases and cancer is crucial because it can form the basis for precision cancer prevention. Diverse and increasingly sophisticated therapeutic options to restore lysosomal functions are currently available or being developed. Future clinical trials of these agents guided by individuals’ variant profiles may pave a new path toward personalized cancer prevention and treatment.
Code availability
The R code used for statistical analysis is available from the first author or the corresponding author upon request.
References
Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471–486.
Platt FM. Sphingolipid lysosomal storage disorders. Nature. 2014;510:68.
Wei H, Kim S-J, Zhang Z, Tsai P-C, Wisniewski KE, Mukherjee AB. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum Mol Genet. 2008;17:469–477.
Jung J, Cho KJ, Naji AK, et al. HRAS‐driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019;20:e46685.
Leusink FK, Koudounarakis E, Frank MH, Koole R, van Diest PJ, Willems SM. Cathepsin K associates with lymph node metastasis and poor prognosis in oral squamous cell carcinoma. BMC Cancer. 2018;18:385.
Petricevic SJ, Pavlovic A, Capkun V, Becic K, Durdov MG. Cathepsin K expression in melanoma is associated with metastases. Histol Histopathol. 2017;32:711–716.
Machado E, White-Gilbertson S, van de Vlekkert D, et al. Regulated lysosomal exocytosis mediates cancer progression. Science Adv. 2015;1:e1500603.
Uemura T, Shiozaki K, Yamaguchi K, et al. Contribution of sialidase NEU1 to suppression of metastasis of human colon cancer cells through desialylation of integrin β4. Oncogene. 2009;28:1218.
Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J. 2007;401:1–11.
Shachar T, Bianco CL, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH. Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov Disord. 2011;26:1593–1604.
Arends M, van Dussen L, Biegstraaten M, Hollak CE. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br J Haematol. 2013;161:832–842.
Cassiman D, Claes K, Lerut E, et al. Bilateral renal cell carcinoma development in long-term Fabry disease. J Inherit Metab Dis. 2007;30:830–831.
Wang RY, Bodamer OA, Watson MS, Wilcox WR. Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med. 2011;13:457–484.
Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. 2007;9:34–45.
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Scriver CR. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001.
Boustany R-MN. Lysosomal storage diseases—the horizon expands. Nat Rev Neurol. 2013;9:583–598.
Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–565.
Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2015;44(D1):D862–D868.
Stenson PD, Mort M, Ball EV, Shaw K, Phillips AD, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9.
Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13:762–775.
Storey JD. A direct approach to false discovery rates. J R Stat Soc Series B Stat Methodol. 2002;64:479–498.
The International Cancer Genome Consortium, Hudson TJ, Anderson W, et al. International network of cancer genome projects. Nature. 2010;464:993.
Easton DF, Pooley KA, Dunning AM, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087.
Yeager M, Orr N, Hayes RB, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645.
Buckley AR, Standish KA, Bhutani K, et al. Pan-cancer analysis reveals technical artifacts in TCGA germline variant calls. BMC Genomics. 2017;18:458.
Waddell N, Pajic M, Patch A-M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501.
Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405.
Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806.
Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011;476:163–169.
Van Hove JLK, Wevers RA, Van Cleemput J, et al. Late-onset visceral presentation with cardiomyopathy and without neurological symptoms of adult Sanfilippo A syndrome. Am J Med Genet A. 2003;118A:382–387.
Trudel S, Trécherel E, Gomila C, et al. Oxidative stress is independent of inflammation in the neurodegenerative Sanfilippo syndrome type B. J Neurosci Res. 2015;93:424–432.
Rylova SN, Amalfitano A, Persaud-Sawin D-A, et al. The CLN3 gene is a novel molecular target for cancer drug discovery. Cancer Res. 2002;62:801–808.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30.
Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378:607–620.
Klein AP. Identifying people at a high risk of developing pancreatic cancer. Nat Rev Cancer. 2013;13:66–74.
Hollak CEM, Wijburg FA. Treatment of lysosomal storage disorders: successes and challenges. J Inherit Metab Dis. 2014;37:587–598.
Acknowledgements
This work was supported by the Seoul National University R&DB Foundation, funded by the Ministry of Health & Welfare, Republic of Korea (HI18C1876); Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (HI14C2399); and the computing resources of Global Science experimental Data hub Center (GSDC) and Korea Research Environment Open NETwork (KREONET). The authors are grateful to Jan O. Korbel, a cochair of the PCAWG germline working group, for providing advice on this study. The manuscript was edited by native English-speaking experts associated with BioScience Writers LLC, Houston, TX.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Shin, J., Kim, D., Kim, HL. et al. Oncogenic effects of germline variants in lysosomal storage disease genes. Genet Med 21, 2695–2705 (2019). https://doi.org/10.1038/s41436-019-0588-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0588-9
Keywords
This article is cited by
-
Genetic assessment of pathogenic germline alterations in lysosomal genes among Asian patients with pancreatic ductal adenocarcinoma
Journal of Translational Medicine (2023)
-
Mortality in patients with alpha-mannosidosis: a review of patients’ data and the literature
Orphanet Journal of Rare Diseases (2022)
