
Abstract
Purpose
Clinically relevant variants exhibit a wide range of penetrance. Medical practice has traditionally focused on highly penetrant variants with large effect sizes and, consequently, classification and clinical reporting frameworks are tailored to that variant type. At the other end of the penetrance spectrum, where variants are often referred to as “risk alleles,” traditional frameworks are no longer appropriate. This has led to inconsistency in how such variants are interpreted and classified. Here, we describe a conceptual framework to begin addressing this gap.
Methods
We used a set of risk alleles to define data elements that can characterize the validity of reported disease associations. We assigned weight to these data elements and established classification categories expressing confidence levels. This framework was then expanded to develop criteria for inclusion of risk alleles on clinical reports.
Results
Foundational data elements include cohort size, quality of phenotyping, statistical significance, and replication of results. Criteria for determining inclusion of risk alleles on clinical reports include presence of clinical management guidelines, effect size, severity of the associated phenotype, and effectiveness of intervention.
Conclusion
This framework represents an approach for classifying risk alleles and can serve as a foundation to catalyze community efforts for refinement.
Similar content being viewed by others


INTRODUCTION
Genetic variants that contribute to disease lie on a spectrum from rare alleles with large effect sizes to more common alleles with small effect sizes.1,2,3 Genetic diseases have historically been categorized as either Mendelian (i.e., caused by variants at a single locus that segregate with a recognizable pattern within families) or complex (i.e., caused by a combination of multiple variants and environmental factors with some degree of heritability that does not follow a clear inheritance pattern).4 Accordingly, Mendelian variants are identified by studying affected families, whereas variants associated with common and complex disease are identified through association studies involving large populations of unrelated individuals.4 This traditional, binary classification of disease was appropriate when the field focused on the most penetrant and severe heritable conditions but does not adequately describe the known landscape of heritable conditions today. It has long been known that many pathogenic variants do not always lead to disease when present in an individual (i.e., show reduced penetrance), but we are only now determining the enormous gradient of penetrance associated with variants that cause or contribute to genetic disease.
While the extreme ends of the penetrance and effect size spectrums are well described, clinically relevant variants in the “gray zone” between Mendelian and complex inheritance are ill-defined with regard to terminology, classification, and clinical reportability. These variants are found in the population more commonly than classic Mendelian alleles and can be inherited in sometimes recognizable familial patterns. Such variants are identified by both Mendelian case studies and population-based association studies and tend to be described using terminology depending on which study initially identified them. Mendelian frameworks refer to these variants as “low-penetrance variants” and complex disease studies describe them as “risk variants” or “risk alleles.” For simplicity, we will use the term “risk allele” throughout this paper.
Some clinically significant risk alleles are well characterized and have long been included on clinical reports, but the lack of consensus terminology and interpretation criteria for this variant type has led to inconsistent classification.5 Additional confusion occurs when risk alleles have frequencies in population databases that meet Mendelian classification standards to be classified as “benign.”6,7 A well-known example is the F5 p.Arg534Gln variant (factor V Leiden), which is present in 3% of European alleles in gnomAD (https://gnomad.broadinstitute.org/variant/1-169519049-T-C, accessed 19 Ocotber 18) and has been submitted to ClinVar by 9 laboratories as “pathogenic” (n = 4), “benign” (n = 1), and “risk variant” (n = 4) (ClinVar ID 642, accessed 19 October 2018). Such divergent classifications can create confusion for patients and clinical practitioners.
With costs of genomic sequencing rapidly decreasing, large population-based studies are increasingly identifying such risk alleles.8,9 Additionally, genomic testing is beginning to be offered to healthy individuals10,11 and there is increasing interest in returning these variants on clinical reports. As such, it is critical that the community defines frameworks for evaluating the validity of evidence supporting the role of risk alleles in disease and develops terminology to clearly distinguish these from variants that cause highly penetrant, Mendelian disease.
Furthermore, consensus is needed regarding what level of evidence warrants inclusion of risk alleles on clinical reports. While the scientific validity of the associated risk has to be the foundation, additional factors need to be considered to balance clinical utility and possible risks for unnecessary medical action. Some risk alleles have clear actionability, including those recognized by specific recommendations from professional societies. For example, the p.Ile1307Lys variant in the APC gene is associated with increased risk for developing colorectal cancer, especially in the Ashkenazi Jewish population. The 2018 National Comprehensive Cancer Network (NCCN) guidelines recommend that unaffected individuals with this variant who are lacking family history of colorectal cancer begin colonoscopy screening at age 40, 10 years earlier than the general population (NCCN Genetic/Familial High-Risk Assessment: Colorectal Version 1.2018, https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf; accessed 1 October 2018). In contrast, reporting a variant that confers risk for a rare condition for which no effective preventive measures exist may require different consenting and counseling procedures, as the probability of developing disease and the severity of the impact may be difficult to convey and comprehend.
We compiled a set of representative risk alleles to define data elements that can be used to express the varying degrees of confidence in their ability to contribute to genetic disease. We propose a classification framework that is conceptually similar to the widely used American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) classification system for germline Mendelian disease7 and discuss which criteria may influence inclusion of such variants on clinical genetic reports. This work is intended to catalyze discussion in the genetics community and serve as a basis for refinement and standardization.
MATERIALS AND METHODS
Inclusion criteria
Variants were required to have at least one statistically significant association with a clinically relevant phenotype, as opposed to physical traits such as eye color and height.
Variant set
Variants were selected from internal databases of two clinical testing laboratories (Laboratory for Molecular Medicine and Veritas Genetics) as well as public databases such as ClinVar. Selected variants for review ranged from single variants, multiple variants forming a haplotype, variants conferring risk through compound heterozygosity, and digenic risk variant combinations.
Curation process and classification criteria
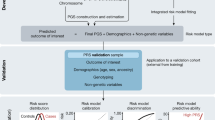
Evidence was gathered systematically using structured data collections forms (Supplementary Figure 1). The proposed framework was applied to the variant set and refined to arrive at a final version (Fig. 1). Variant classifications were performed by two independent curators, reviewed by ABMGG-certified clinical molecular geneticists, and finalized after reaching consensus with the entire group.

Decision-making framework for the classification of risk alleles. LoF loss of function.
Metrics for establishing reportability
We identified criteria for guiding decision-making on reportability of risk alleles based on the final classification level of the variant–disease association and clinical information about the disease that is associated with the variant in question, such as disease prevalence and severity, as well as effectiveness and risk of intervention. We extracted the information from PubMed literature searches, Genetics Home Reference (https://ghr.nlm.nih.gov/), the Centers for Disease Control and Prevention (https://www.cdc.gov/), ACMG Technical Standards and Guidelines (http://www.acmg.net/ACMG/Medical-Genetics-Practice-Resources/Technical_Standards_and_Guidelines.aspx), Clinical Genome Resource Actionability Work Group documents (https://www.clinicalgenome.org/curation-activities/clinical-actionability/the-process/), disease-specific databases (e.g., https://www.nccn.org/professionals/physician_gls/recently_updated.aspx,) and other relevant publications.12,13,14
RESULTS
Classification framework
Classifying any type of variant in a clinical setting requires careful evaluation of the quality of the associated data, aggregation of available evidence, and application of criteria to establish the likelihood with which this evidence predicts the outcome. The following sections describe a proposed framework for assessing and classifying risk alleles using the terminology initially suggested by the ACMG/AMP guidelines for interpretation of germline variants.7 We focused on general steps and concepts (Fig. 1 and sections below) to provide a basis for community iteration and refinement.
Step 1: assessment of study design and data quality
Characteristics of well-designed and reliable association studies have been published15,16,17 and include large, race-matched and well-phenotyped case and control cohorts, application of statistical correction for multiple hypothesis testing, application of a rigorous threshold for statistical significance, and calculation of odds ratios or relative risks as a measure of effect size. We considered any study that reported statistically significant results (p < 0.05) and excluded those reporting effect sizes where the confidence interval included 1.
Step 2: considerations surrounding associated phenotypes
Medical literature often reports association of a variant across a range of phenotypes. While some represent distinct clinical entities, others represent endophenotypes and deciding which studies should be combined can be challenging. This is now a well-recognized phenomenon and early guidance is available to train clinical variant curation professionals (https://www.clinicalgenome.org/site/assets/files/9703/lumping_and_splitting_guidelines_gene_curation_final.pdf; accessed 1 October 2018). While this problem affects variants across the full range of the genetic penetrance spectrum, it is particularly common among genetic association studies.18 We intentionally took a very conservative approach to avoid overclassification of variants. Generally, only studies reporting an association with the full disease or its predominant clinical features were included. For example, for the APOE e4 allele we included studies that demonstrated an association with Alzheimer disease but not association with aggression or depression in Alzheimer patients nor with disease progression once an Alzheimer diagnosis was made.
There can be significant ambiguity as to what defines the disease state. This is particularly pronounced for disorders whose primary defect is a biochemical imbalance, which results in clinical features only when exceeding a threshold. For these disorders, we only considered studies reporting an association with clinically evident phenotypes. For example, hereditary hemochromatosis caused by variants in the HFE gene leads to elevated transferrin levels, which can manifest with symptoms of end-stage organ damage secondary to iron storage. Because elevated serum transferrin alone can have different causes,19 we considered only studies reporting an association with the clinical endpoint (e.g., liver disease).
Step 3: data elements and classification criteria
The strength of a variant–disease association was assigned one of three categories using terminology akin to those commonly used for Mendelian disease and suggested in the ACMG/AMP interpretation guidelines:7 “established risk allele,” “likely risk allele,” or “uncertain risk allele.” Because a large number of association studies fail to replicate,20 highest emphasis was placed on meta-analyses and multiple, independent studies confirming the originally reported association. Additional data elements (such as functional data) were given modifying weight.
Number and types of studies reporting an association
To classify a variant as an “established risk allele” for a condition, we suggest a minimum of one robust meta-analysis or multiple independent case–control studies that each meet all criteria for well-designed studies outlined above. A “likely risk allele” classification requires less evidence and we suggest at least two independent case–control studies showing a statistically significant association with the phenotype of interest. When multiple studies report conflicting results, a “likely risk allele” classification can still be reached when the clear majority are concordant with regard to significance and effect size. Another scenario qualifying for a “likely risk allele” classification is a single, large study of high quality with data from multiple sites.
All other scenarios result in an “uncertain risk allele” classification as a baseline, which can be modified when other supporting evidence, such as functional data, is available. Common examples for “uncertain risk allele” classifications include a single, unreplicated case–control study or replicated associations derived from overlapping cohorts or solely from very small studies. Case–control studies that have already been included in meta-analyses are not individually reviewed and double counted as replication.
Functional data
Evidence demonstrating a direct effect on protein function was given supporting weight, allowing for adjustment of the classification category. Validity, relevance, and reproducibility of the functional data were taken into consideration as recommended by ACMG/AMP guidelines.7 Only strong functional data was allowed to be used in this fashion.7,21 Generally, this included only data from variant-specific in vivo models recapitulating the associated human phenotype or reliable enzymatic assays performed in relevant in vitro systems. Functional evidence of an effect on protein function can provide confidence in disease association and a distinct causal role for the variant, rather than an indirect effect through genetic linkage. In contrast, functional data showing no effect on protein/gene function was not used to downgrade the classification when association study results clearly support a “likely” or “established” risk allele classification as the signal can always be due to another variant that is in linkage disequilibrium. For example, even though in vitro functional studies demonstrated no effect on protein function, the p.Asn34Ser variant in SPINK1 was classified as an “established risk allele” based on evidence from two meta-analyses and one large case–control study (Table 1).
Loss-of-function variant considerations
Most risk alleles are common in the general population, which provides enough statistical power to establish an association with disease. In some instances, it may be possible to assign risk to a class of variants regardless of whether additional published evidence exists. For example, the ACMG/AMP Mendelian classification framework allows assigning substantial weight to a novel loss-of-function (LOF) variant provided that LOF is an established mechanism of disease for the gene. This concept can be extended to genes that are overall associated with lower penetrance. An example is the CHEK2 gene, where LOF appears to be associated with a level of risk for developing of cancer that may be more adequately expressed using the risk framework.22 The most prominent cancer susceptibility variant in this gene is a LOF variant (c.1100delC) that leads to a 37% lifetime risk of cancer, which reaches a level where use of the Mendelian “pathogenic” classification under an autosomal dominant cancer susceptibility framework may be more appropriate.23 However, because the CHEK2 gene is not as well studied as other cancer susceptibility genes, it may be more prudent to describe novel LOF variants using the risk framework and elevate them to a Mendelian classification when data is available that conveys more certainty about the clinical outcome.
Application of the framework
We applied this framework to a set of 33 variants in 22 genes that met characteristics to potentially be classified as risk alleles. Variants and alleles were assessed individually for disease associations across all zygosity states. Data was available to make classifications for 19 heterozygous variants, 9 homozygous variants, and 5 compound or double heterozygous variants. A summary of the criteria met and resulting classifications are listed in Table 1 with additional detail on each classification provided in Supplementary Table 1.
Reporting considerations
Deciding whether or not to return risk alleles in clinical genetic testing can be challenging as the absolute risk and the clinical utility of disease associations are often not as clear as they are for Mendelian disease variants. We defined criteria we believe should be taken into consideration for reporting decisions.
The base criterion for clinical reporting is the scientific validity of the associated risk, which is expressed by the classification category. In our opinion, the clinical utility of returning variants with reported but unconfirmed disease associations (i.e., “uncertain risk alleles”) is low and we propose restricting reporting to “established” and “likely” risk alleles. This is similar to common practice for Mendelian testing, where predictive reports (secondary findings) are restricted to “likely pathogenic” and “pathogenic” variants.9,24 However, while a likely or established risk allele classification constitutes a necessary criterion, it is not sufficient. Below and in Fig. 2 we describe additional criteria that should be considered.

Routing logic for inclusion of risk alleles on clinical reports.
A major consideration for reporting is availability of clinical management guidelines issued by expert groups or professional societies. Variants classified as established or likely risk allele with such guidelines were considered candidates to include on clinical reports. For other risk alleles, we discuss five additional criteria that could be combined into an “impact score” reflecting their overall clinical importance: (1) effect size, (2) disease prevalence, (3) disease severity, (4) effectiveness of intervention, and (5) risk associated with action/intervention. The scores for (3)–(5) were based upon the semiquantitative metrics put forth by the ClinGen Actionability Working Group.13
To arrive at this impact score, we assigned a numerical value from 0 to 3 to each criterion (3 having the greatest weight). We calculated combined scores for three illustrative risk alleles (p.Asp85Asn [KCNE1], p.Glu318Lys [MITF], and p.Val210Ile [PRNP]) (Table 2, Fig. 3). Further consideration by the broader community will be needed to refine this approach and determine a universal threshold for reportability.

Reportability scores. Radar charts visualize five reportability criteria for three variants.
As genomic testing is increasingly administered in an elective fashion, personal utility and testing scenario (diagnostic versus predictive) should also be considered. Indication-based testing may include risk alleles related to the phenotype depending upon the benefits and hazards of reporting, such as overinterpretation of the risk. Predefining this criterion allows for risk alleles to be evaluated during the test design stage. Additionally, reporting risk alleles as secondary findings from genomic screening may require a higher bar for inclusion due to the added uncertainty of interpreting such findings in individuals who do not present with features of the associated disease. This is similar to current practice for Mendelian disease testing, where variants of unknown significance (VUS) are commonly reported in a diagnostic setting but are typically not returned in predictive testing scenarios.
Considerations for communicating the significance of risk on a clinical report
Similar to what is customary for reporting Mendelian disease variants, risk alleles should be accompanied by a summary of all evidence supporting a classification. If an association study reports a statistically significant odds ratio or other statistical measure, these values along with confidence intervals and p values should be stated or summarized. In addition, as statistical measures derived from this framework do not represent absolute risks, care needs to be taken to communicate this clearly and avoid overinterpretation by the recipient. Furthermore, the absolute risk increase attributable to the presence of a risk allele is a function of both the effect size of the risk allele as well as the prevalence of the associated disease. When reporting risk alleles associated with rare diseases, one must consider that the absolute risk increase may be small and clinically insignificant despite a large effect size. In contrast, when reporting risk alleles in common disease, absolute risk increases resulting from risk alleles with modest effect sizes, as expressed in odds ratios, may be large and clinically significant.
Additionally, it is important to indicate when studies are limited to populations of a specific ancestry, as many risk variants are identified within specific populations, typically Caucasian. Since the risk variant may only be in linkage disequilibrium with the causative variant, the associated risk may not translate to a population with different linkage disequilibrium architecture. Furthermore, when conveying absolute risk, there may be marked differences in different populations due to differences in baseline risk for the disease. It is, therefore, prudent to consider the reported or computed ancestry of the individual when conveying risk on the report.
For risk alleles that are classified as “likely” or “established” but for which contradictory (i.e., negative) functional data exists, a clarifying statement is important to avoid misinterpretation and dismissal of risk by the recipient of the report. Included in the evidence should be language that mentions the possibility that the variant may not be the causal variant but may be linked to another, unidentified variant or that the function that was assessed by the assay may not reflect the protein function that is relevant for disease expression.
A step-by-step application of the framework and interpretive summary for one risk variant are provided in Supplementary Document 1.
DISCUSSION
The medical community has long been aware of variants that fall into the gray zone between rare, highly penetrant variants and variants contributing to common or complex disease. While the extreme ends of this penetrance gradient are clinically well defined, little guidance exists for variants with significantly reduced penetrance but effect sizes that warrant consideration in a clinical setting. As was the case for Mendelian variants, the lack of standards has led to discordance in how these variants are evaluated and labeled, which may ultimately have negative consequences if they are classified as “benign” and not reported to the patient. As genomic testing is shifting toward exome and genome sequencing, large studies are increasingly revealing risk alleles associated with medically relevant conditions. Simultaneously, the rise of elective genome screening in healthy individuals is increasing the demand to return such variants on clinical reports.
To address the emerging need for guidance, we developed a proposed first framework to systematically evaluate the scientific validity of reported risk allele associations. We define the data elements that should be evaluated and suggest a method to assign clinical classifications. The utility of such frameworks is well established and is known to lead to harmonization between clinical laboratories. This is most recently evidenced by the enormous impact of the ACMG/AMP variant classification framework for Mendelian variants,7 which has led to an impressive amount of community harmonization.5,25,26 Additionally, we raise the question as to which factors should guide inclusion of risk alleles on clinical reports. Scientific validity is the minimum requirement, but even more than for Mendelian disorders, the risk for overinterpretation by the recipient and the resultant possibility for causing harm and anxiety has to be carefully considered for risk alleles.
Our approach was deliberately conservative and designed to raise concepts rather than suggest prescriptive guidance. The framework will require iteration via community input and ultimately professional society recommendations. Whether or not to return risk alleles will also be impacted by the testing scenario. We predict that the “bar” for including risk alleles in a healthy/elective testing scenario may be more stringent than in a diagnostic setting where one may include uncertain risk alleles relevant to the indication, similar to what is common practice in traditional, Mendelian testing.9 Finally, the framework we present here can be extended to protective alleles, which have been largely ignored in a traditional clinical testing setting but are expected to increase in demand as genomic testing is further expanded to the prediction of disease risk.
Further thought will also be needed in a number of important areas:
- (1)
It is not trivial to decide how to construct clinical reports that contain variants across the risk/penetrance spectrum. Separating risk alleles from highly penetrant, Mendelian variants is an option but in complex reports actionability considerations that are associated with some risk alleles may be then ignored. An example is p.Ile1307Lys in APC for which NCCN management guidelines recommend a modified colorectal cancer screening protocol. Such variants may need to be grouped with other actionable variants.
- (2)
Consensus is needed on how to accurately communicate the uncertainty surrounding any quantitative risk estimate. It will be important to avoid large discrepancies in risk estimates between clinical laboratories, which have previously plagued the direct-to-consumer testing space.27 Consensus lists of reportable risk alleles would further help harmonize clinical reporting practices between laboratories.
- (3)
The penetrance threshold separating variants that should be classified by the Mendelian framework from risk alleles will need to established and this may differ between disease areas within clinical genetics. For example, cancer predisposition testing, which includes the CHEK2 c.1100delC variant, has a longstanding history of classifying variants within the Mendelian framework despite incomplete penetrance for many variant–cancer type associations. In our opinion, as a general rule when penetrance data is unavailable, variants whose disease association has been demonstrated through only segregation analysis within affected families should be classified within the Mendelian framework whereas variants identified in association studies or case–control cohorts of unrelated individuals should be classified as risk alleles.
While the set of variants examined was small, we collected initial impressions regarding the amount of time laboratories will need to budget for classifying risk alleles. In our experience, the relative simplicity of the risk framework is counterbalanced by the need to evaluate many publications, leading to an overall time that is similar to what is typically budgeted for Mendelian variants with publications (2–3 hours). It is worth noting that risk alleles can often be assessed during product development and then do not impact postlaunch clinical operations.
Future challenges in developing clinically relevant quantitative risk estimates include (1) calculating the patient’s pretest risk based on demographic, clinical, environmental, and other genetic variables; (2) selecting and integrating the published data used to calculate the updated risk estimate; (3) incorporating the effects of environmental variables; (4) updating the risk model in an accurate and transparent manner; and (5) appropriately expressing uncertainty surrounding the calculated risk estimate in the clinical report.
Our work serves as a starting point for the structured classification and reporting of risk alleles in clinical genetic testing reports. We believe that the framework is generalizable as it relies on concepts that have been established in ranking the quality and replicability of association studies. However, we acknowledge that it has thus far only been tested on a very small number of variants, making it likely that rules will need to be refined and expanded over time. We look forward to continued advancement and harmonization on this subject within the broader clinical genetics community. A first step toward community engagement is now underway via a working group assembled by the Clinical Genome Resource (ClinGen) with the goal of producing a classification methodology with broad community input.
References
Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753.
Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17:1520–1528.
Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk of complex disease. Curr Opin Genet Dev. 2008;18:257–263.
Antonarakis SE, Chakravarti A, Cohen JC, Hardy J. Mendelian disorders and multifactorial traits: the big divide or one for all? Nat Rev Genet. 2010;11:380–384.
Harrison SM, Dolinksy JS, Chen W, et al. Scaling resolution of variant classification differences in ClinVar between 41 clinical laboratories through an outlier approach. Hum Mutat. 2018;39:1641–1649.
Duzkale H, Shen J, Mclaughlin H, et al. A systematic approach to assessing the clinical significance of genetic variants. Clin Genet. 2013;84:453–463.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Khan R, Mittelman D. Consumer genomics will change your life, whether you get tested or not. Genome Biol. 2018;19:120.
Lu JT, Ferber M, Hagenkord J, et al. Evaluation for genetic disorders in the absence of a clinical indication for testing: elective genomic testing. J Mol Diagn. 2019;21:3–12.
Holm IA, Agrawal PB, Ceyhan-Birsoy O, et al. The BabySeq project: implementing genomic sequencing in newborns. BMC Pediatr. 2018;18:225.
Vassy JL, Christensen KD, Schonman EF, et al. The impact of whole-genome sequencing on the primary care and outcomes of healthy adult patients: a pilot randomized trial. Ann Intern Med. 2017;167:159–169.
Berg JS, Foreman AKM, O’Daniel JM, et al. A semiquantitative metric for evaluating clinical actionability of incidental or secondary findings from genome-scale sequencing. Genet Med. 2016;18:467–475.
Hunter JE, Irving SA, Biesecker LG, et al. A standardized, evidence-based protocol to assess clinical actionability of genetic disorders associated with genomic variation. Genet Med. 2016;18:1258–1268.
Webber EM, Hunter JE, Biesecker LG, et al. Evidence-based assessments of clinical actionability in the context of secondary findings: updates from ClinGen Actionability Working Group. Hum Mutat. 2018;39:1677–1685.
Minelli C, Thompson JR, Abrams KR, Thakkinstian A, Attia J. The quality of meta-analyses of genetic association studies: a review with recommendations. Am J Epidemiol. 2009;170:1333–1343.
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151:264–269.
Stack CB, Gharani N, Gordon ES, Schmidlen T, Christman MF, Keller MA. Genetic risk estimation in the Coriell Personalized Medicine Collaborative. Genet Med. 2011;13:131–139.
Bush WS, Moore JH. Chapter 11: genome-wide association studies. PLoS Comput Biol. 2012;8:e1002822.
Wood JC. Guidelines for quantifying iron overload. Hematol Am Soc Hematol Educ Program. 2014;2014:210–215.
Kraft P, Zeggini E, Ioannidis JPA. Replication in genome-wide association studies. Stat Sci. 2009;24:561–573.
Macarthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476.
Apostolou P, Papasotiriou I. Current perspectives on CHEK2 mutations in breast cancer. Breast Cancer (Dove Med Press). 2017;9:331–335.
Weischer M, Bojesen SE, Ellervik C, Tybjærg-Hansen A, Nordestgaard BG. CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: meta-analyses of 26,000 patient cases and 27,000 controls. J Clin Oncol. 2008;26:542–548.
Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574.
Amendola LM, Berg JS, Horowitz CR, et al. The Clinical Sequencing Evidence-Generating Research Consortium: integrating genomic sequencing in diverse and medically underserved populations. Am J Hum Genet. 2018;103:319–327.
Lebo MS, Zakoor K-R, Chun K, et al. Data sharing as a national quality improvement program: reporting on BRCA1 and BRCA2 variant-interpretation comparisons through the Canadian Open Genetics Repository (COGR). Genet Med. 2018;20:294–302.
Imai K, Kricka LJ, Fortina P. Concordance study of 3 direct-to-consumer genetic-testing services. Clin Chem. 2011;57:518–521.
Acknowledgements
We acknowledge Carrie Blout, Megan Maxwell, and the team at Genomes2People for initial feedback on the classification of risk variants. We also acknowledge the MilSeq project for evaluating the return of risk alleles in a genomic screening context.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
E.Q. and B.F. are employed by Veritas Genetics, a company that offers clinical elective genome sequencing. M.S.L. and H.M.-S. work for the Laboratory for Molecular Medicine, a nonprofit fee-for-service clinical laboratory performing genetic testing and genomic screening. R.J.S works at the Children’s Hospital Los Angeles Center for Personalized Medicine, a nonprofit fee-for-service clinical laboratory performing genetic testing. O.S.-C. is currently employed by Janssen: Pharmaceutical Companies of Johnson & Johnson. D.H. is currently employed by Tempus Labs, a company offering clinical genetic testing.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Senol-Cosar, O., Schmidt, R.J., Qian, E. et al. Considerations for clinical curation, classification, and reporting of low-penetrance and low effect size variants associated with disease risk. Genet Med 21, 2765–2773 (2019). https://doi.org/10.1038/s41436-019-0560-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0560-8
Keywords
This article is cited by
-
Genetically transitional disease: conceptual understanding and applicability to rheumatic disease
Nature Reviews Rheumatology (2024)
-
Calculating variant penetrance from family history of disease and average family size in population-scale data
Genome Medicine (2022)
-
Development of a clinical polygenic risk score assay and reporting workflow
Nature Medicine (2022)
