
Abstract
Purpose
Mediator is a multiprotein complex that allows the transfer of genetic information from DNA binding proteins to the RNA polymerase II during transcription initiation. MED12L is a subunit of the kinase module, which is one of the four subcomplexes of the mediator complex. Other subunits of the kinase module have been already implicated in intellectual disability, namely MED12, MED13L, MED13, and CDK19.
Methods
We describe an international cohort of seven affected individuals harboring variants involving MED12L identified by array CGH, exome or genome sequencing.
Results
All affected individuals presented with intellectual disability and/or developmental delay, including speech impairment. Other features included autism spectrum disorder, aggressive behavior, corpus callosum abnormality, and mild facial morphological features. Three individuals had a MED12L deletion or duplication. The other four individuals harbored single-nucleotide variants (one nonsense, one frameshift, and two splicing variants). Functional analysis confirmed a moderate and significant alteration of RNA synthesis in two individuals.
Conclusion
Overall data suggest that MED12L haploinsufficiency is responsible for intellectual disability and transcriptional defect. Our findings confirm that the integrity of this kinase module is a critical factor for neurological development.
Similar content being viewed by others


INTRODUCTION
Mediator is a key regulator of gene expression involved in cell growth, homeostasis, development, and differentiation.1 Mediator is a large multiprotein complex composed of four different modules (Kinase, Head, Middle, and Tail), which conveys essential information from proteins bound at DNA response elements to the basal transcription machinery located around the transcription initiation site.2,3 Dysfunction of the transcription machinery components, including Mediator, has been shown to elicit a range of effects on cell states giving rise to diverse disorders including developmental delay and intellectual disability. Variants in Mediator subunits are associated with a wide range of genetic disorders, most of them exhibiting neurological disabilities.4 Genetic and biochemical studies have established the Mediator Kinase module as a major ingress of developmental and oncogenic signaling through the three other modules constituting the core component, and much of its function derives from its resident CDK8 kinase activity likely regulated by its association with MED13, MED12, and Cyclin C (CycC) subunits. Recent studies have shown that the kinase module can also encompass CDK19, MED12L, and MED13L, which are paralogs of CDK8, MED12, and MED13 respectively.5 Little is known about their biological roles, but each of these proteins appears to assemble in a mutually exclusive fashion with its paralog.
Germline variants of MED12 have already been found in several genetic disorders associated with X-linked intellectual disability, such as Opitz–Kaveggia syndrome (also named FG syndrome),6,7,8 Lujan syndrome (p.N1007S),9 and Ohdo syndrome.10,11 All of these MED12-related disorders exhibit defects in gene expression.12 Variants in CDK19, MED13, and MED13L have also been associated with neurodevelopmental disorders,13,14,15 likely due to defects in gene transcription.
Here, we report a series of individuals sharing variants involvingMED12L (mediator complex subunit 12 like) (OMIM 611318) recruited through an international collaboration and identified by array comparative genomic hybridization (aCGH), exome or genome sequencing. Our attention focused on the effect of MED12L on gene expression, studying transcription machinery of two individuals’ fibroblasts. Whereas CDK19, MED12L, and MED13L are now all linked to neuronal and developmental disorders, their basic biological importance relative to CDK8, MED12, or MED13 remains unclear.
MATERIALS AND METHODS
Individual recruitment
The compilation of this case series resulted from an international collaborative effort among Centre Hospitalier Universitaire (CHU) Nantes, Strasbourg University, CHU Caen, Baylor Genetics Laboratories (BG), GeneDx, HudsonAlpha Institute for Biotechnology, and University of Oklahoma School of Medicine. It was also partly facilitated by the web-based tools GeneMatcher and DECIPHER.16,17 All participants were clinically assessed by at least one clinical geneticist from one of the participating centers. The study was approved by the CHU de Nantes ethics committee (number CCTIRS: 14.556). Consent for the publication of photographs (Fig. 1) was obtained for individuals 1, 2, and 5.

Photographs and brain magnetic resonance images (MRI) of individuals with variants inMED12L. a Individual 1: unilateral ptosis with iris coloboma, hypertelorism, sparse eyebrows, downslanted palpebral fissures, bulbous nasal tip. b Individual 2: prominent nasal bridge, short philtrum, everted lower lip, small mouth. c Individual 5: deep-set eyes, bulbous nasal tip, thin upper lip, triangular face. Brain MRI shows mildly hypoplastic corpus callosum, which is foreshortened with a small splenium.
Molecular analysis
Institutional review board–approved written informed consent was obtained for all subjects. DNA was extracted from leukocytes according to standard procedures.
Copy-number variants (CNVs) were found by microarray-based comparative genomic hybridization (array CGH). Different array platforms were used for genomic copy-number analyses, which were carried out according to manufacturers’ recommendations (Agilent CGH Microarray 60 K or 180 K [Agilent Technologies, Santa Clara, CA]). Chromosomal rearrangements were confirmed by fluorescence in situ hybridization (FISH) with various specific probes on chromosome preparations from leukocyte cultures or by quantitative polymerase chain reaction (PCR) using standard protocols. Several different oligonucleotide probes were used to try to sequence the breakpoints of the CNV carried by individual 1. Parental testing was performed when DNA samples were available. Genomic positions are relative to human genome build GRCh37/hg19. The CNVs were submitted to the DECIPHER database.
Single-nucleotide variants (SNV) were all identified by exome or genome sequencing. The protocols used by each participating center have been detailed elsewhere.18,19 Reads were aligned to the human reference genome sequence (National Center for Biotechnology Information [NCBI] build 37.3, hg19). For SNVs, segregation analysis was made by direct sequencing and then performed in the families to confirm inheritance when parental DNA samples were available. The variants were submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).
RRS assay
Recovery of RNA synthesis (RRS) was evaluated on primary fibroblast cultures using fluorescent nonradioactive assay as described previously.20 RRS evaluates the transcription-coupled repair pathway (TC-NER). Cells were plated on coverslips in 6-well plates at a confluence of 7 × 104 cells per well. After 2 days in culture, cells were washed with phosphate-buffered saline (PBS), followed by irradiation with a range of UV-C doses (6-12-20 J/m2). The nonirradiated plates acted as references. After UV irradiation, cells were incubated for 23 hours for RNA Synthesis recovery in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with fetal bovine serum. Then, after washing with phosphate buffered saline (PBS), cells were labeled with 5-ethynyl-uridine (EU; Invitrogen) for 2 hours. Cells were then washed again with PBS, followed by fixation and permeabilization. The last step involved an azide-coupling reaction and DAPI (6′-diaminido-2-phenylindole) staining (Click-iT RNA HCS Assay, Invitrogen). Finally coverslips were washed in PBS and mounted on glass slides with Ibidi Mounting Medium (Biovalley). Photographs of the cells were taken with a fluorescent microscope (Imager.Z2) equipped with a charge-coupled device (CCD) camera (AxioCam, Zeiss). The images were processed and analyzed with the ImageJ software. At least 50 cells were randomly selected, and the average nuclear fluorescent intensity was calculated.
RESULTS
Clinical description
We identified seven affected individuals from seven independent families, including four males and three females. The main clinical features of our cohort are summarized in Table 1 and Fig. 1 and detailed in Supplementary Data.
All individuals had neurological impairment. Intellectual disability/developmental delay was the main feature (7/7). Three individuals had a mild intellectual disability; three had a moderate intellectual disability with mild to severe speech impairment. One individual had a severe intellectual disability with seizures and brain abnormalities. Abnormal behavior was common (6/7) including mild to severe autism spectrum disorder (4/7) and aggressive behavior (4/7). Attention deficit with hyperactivity (3/7), sleep disorder (3/7), and hypotonia (1/7) were also noted. One individual had seizures and another one had staring spells with the electroencephalogram (EEG) within normal limits. Brain magnetic resonance imaging (MRI) was performed in four individuals showing abnormalities for three of them: corpus callosum agenesis or hypoplasia (2/4), and secondary cortical signal abnormality with volume loss of bilateral putamen and globus pallidus (1/4).
Gastrointestinal issues were reported in 5/7 individuals, including chronic constipation (3/7), feeding difficulties (2/7, one individual required a gastrostomy tube placement), gastroesophageal reflux (2/7), and inguinal hernia (1/7). Various other miscellaneous extraneurological features were reported such as congenital malformations (2/7, including iris coloboma and hypospadias), ophthalmologic features (3/7), skeletal abnormalities (2/7), and minor hand anomalies (4/7). Mild facial morphological features were reported in some individuals, but without a recognizable phenotype.
Genetic results
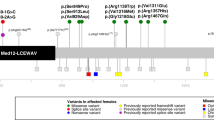
Genomic positions of copy-number variants (CNVs) and single-nucleotide variants (SNVs) identified in MED12L are mapped in Fig. 2.

Mapping of theMED12Llocus. a Genomic position of MED12L variants identified in our series. Upper panel represents single-nucleotide variants. Lower panel represents copy-number variants. b Localization of predicted domains of MED12L protein.
Individuals 1, 2, and 3 harbored CNVs involving MED12L ranging in size from 147 Kb to 460 Kb (DECIPHER accession number: 284908, 280845, and 277489). Individual 1 carried a 460-Kb duplication involving the terminal part of MED12L. Attempts to sequence the breakpoints of the duplication by Sanger failed arguing for a complex rearrangement. Individual 2 carried a 291-Kb deletion involving the terminal part of MED12L. Individual 3 carried a 147-Kb intragenic MED12L duplication. Minimal and maximal coordinates of the CNVs are indicated in Table 1. CNVs were found to be de novo in individuals 1 and 2. Parents were not available for individual 3.
Genome sequencing was performed in individual 4 and exome sequencing in individuals 5 to 7. One frameshift, one nonsense, and two splice variants were identified in MED12L (NM_053002.5, exons are numbered like in NG_021244.1): c.1747dup, p.(Ser583PhefsTer8), c.5992C > T, p.(Gln1998Ter), c.4374-1G > A, c.4686-1G > A (ClinVar accessions: SCV000611598, SCV000853261, SCV000853262, and SCV000853263). Variants were absent from gnomAD, Exome Variant Server (EVS), and 1000 Genome databases. Nonsense variants likely result in the degradation of messenger RNA (mRNA) by nonsense-mediated decay (NMD). Segregation analysis showed that the variants occurred de novo when parents’ DNA was available (individuals 5 and 7).
Lastly, individual 4 harbored a variant of unknown significance inTUBB2B (OMIM 612850) that could not be tested for inheritance or excluded as contributing to the individual overall phenotype (see Table 1).
Functional analysis
To test the functional consequences of the MED12L variants on transcriptional processes, we used the recovery of RNA synthesis (RRS) assay, which is the gold standard diagnostic tool in Cockayne syndrome (CS), another transcription-related human disease with syndromic intellectual disability.21 This assay reflects the global transcriptional activity by measuring the incorporation of fluorescent uridine 24 hours after UV irradiation. In wild-type cells, UV irradiation temporarily halts the transcription of a large subset of genes, and 24 hours later they recover a normal RNA synthesis activity.22 Such an assay is used to partly synchronize cells and enable a standardized assessment of global transcriptional activity. Primary fibroblasts from two individuals withMED12L CNV (individuals 1 and 2) were available for RRS testing and were compared with fibroblasts from a healthy control, from a classical Cockayne individual (carrying pathogenic variants in the ERCC8/CSA gene) and from individuals carrying pathogenic variants in other mediator subunit genes, MED12 andMED13L. Both MED12L cell lines show a moderate but significant decreased RNA synthesis level 24 hours after UV irradiation, as compared with the healthy control, similar to the level observed in a control MED12 cell line (Fig. 3). The control MED13L cell line showed a more severely reduced RNA synthesis level, closer to the classical and the severe transcriptional defect observed in the control Cockayne cell line.22

Recovery of RNA synthesis (RRS) following UV irradiation. Fibroblasts of individuals 1 (△) and 2 (○) show a moderately decreased level of RRS as compared with the normal control cell line (♦), similar to a MED12 mutated cell line (●). A MED13L mutated cell line (x) shows a more severely decreased RRS level, closer to the typical severely decreased level of RRS in a CSA defective cell line (■). Error bars represent standard errors of the mean.
DISCUSSION
Here we described a series of seven individuals presenting with variants (CNVs and SNVs) involving MED12L. Individual 1 carried a partial de novo duplication of MED12L. Despite not being proven by our study, we suspected a complex rearrangement. We also showed that the duplication was associated with a defect in transcriptional activity of fibroblasts. Individual 2 carried a partial deletion with a similar transcriptional defect as for individual 1. Individuals 3, 4, and 5 carried respectively an intragenic duplication, a nonsense variant (localized in exon 39/43), and a frameshift variant with a premature nonsense variant, all predicted to result in an altered mRNA likely eliminated by NMD. Individuals 6 and 7 carried intronic variants predicted to alter splicing resulting in a premature nonsense codon. Most variants were de novo. Unfortunately, segregation was not possible for three patients either because they were adopted (individuals 4 and 6) or because parents’ DNA were not available (individual 3). Functional studies were performed only for two French patients. Fibroblasts were not available for the other individuals. All in all, despite these limitations, those data argue for a haploinsufficiency mechanism leading to transcriptional defect.
All individuals described here showed intellectual disability/developmental delay or speech impairment, sometimes associated with abnormal behavior, corpus callosum agenesis, and mild facial morphological features. No genotype–phenotype correlation could be identified in our series. Individual 1 also harbored a 22q11.2 duplication, which is considered as a risk factor for attention deficit hyperactivity disorder, autism, and intellectual disability with a low penetrance estimated around 10%23. The healthy mother of individual 1 carried the same 22q11.2 duplication. Even if we could not exclude a partial role of this duplication, functional data provided and the severity of intellectual disability tended to suggest that MED12L CNV had a more important role in the phenotype of individual 1.
MED12L contains 43 exons and encodes a protein component of Mediator, which is involved in transcriptional coactivation of nearly all RNA polymerase II–dependent genes.22 MED12L is localized to the nucleus and mainly expressed in brain (www.proteinatlas.org). By homology, three domains are predicted to be common with MED12, including a LCEWAV-domain (AA 283–730), with a conserved sequence motif of unknown function, and a catenin-binding domain (AA 1802–2019), activating the canonical Wnt/β-catenin pathway. The measure of probability of intolerance to loss of function (pLI) score, based on the difference in observed and expected loss-of-function variants in the gene, indicates MED12L is extremely intolerant to heterozygous loss of function (pLI score of 1).24MED12L overlaps with six genes, five of which—P2RY12, P2RY13, P2RY14, GPR171, and GPR87—encode purinergic receptors participating in vascular and immune response to injury, and currently have no known link to neurodevelopment. MED12L also overlaps IGSF10, which encodes an immunoglobin involved in the control of early migration of neurons expressing gonadotropin-releasing hormone. A MED12L variant (rs1554120) was associated (p = 5.25E-06) with cortical thickness of right Heschl’s gyrus (HG) by genome-wide association study.25 HG is a core region of the auditory cortex whose morphology is highly variable across individuals. This variability has been linked to sound perception ability in both speech and music domains. MED12L interacts with NCAPD2 in human.26NCAPD2 encodes a non-SMC (structural maintenance of chromosomes) subunit of the condensin complex and causes autosomal recessive microcephaly.27
Mediator is highly conserved across eukaryotes.22 It links gene-specific transcription activators with the basal transcription machinery. Mediator contains 30 subunits organized into four modules: MED12L is predicted to be a subunit of the kinase module with MED12, MED13, MED13L, CDK8, CDK19, and Cyclin C. The kinase module is reversibly associated with the Mediator core and is involved in transcriptional repression and activation. The function of this module could be mediated by its kinase activity when it is recruited to an upstream activation region or enhancers, or by the mutually exclusive interaction between the kinase module and RNA polymerase II with the Mediator core integrating the preinitiation complex.28,29 The Mediator complex is also involved in chromatin regulation, and mRNA processing.3,30,31
Other Mediator kinase module subunits have already been implicated in human disease, namely MED12, MED13, MED13L, and CDK19. Variants in MED12 have been reported in Opitz–Kaveggia (FG) syndrome, Lujan–Fryns syndrome, and Ohdo syndrome, Maat–Kievit–Brunner type.6,8,9,10 Those three X-linked intellectual disability syndromes are allelic and caused by missense MED12 variants and defects in gene expression. FG syndrome and Lujan–Fryns syndrome share some overlapping features such as dysgenesis of the corpus callosum, relative macrocephaly, a tall forehead, and hypotonia. Chronic constipation or anal anomalies are a frequent finding in individuals with FG syndrome. Males with FG syndrome also have deficits in communication skills despite affability and excessive talkativeness, attention deficit hyperactivity disorder, maladaptive behavior with aggressive behavior, and anxiety.32 Marfanoid habitus and hypernasal speech are related to Lujan–Fryns syndrome. Hyperactivity, aggressive behavior, shyness, and attention-seeking behavior are also common in individuals with Lujan–Fryns syndrome. Consistent facial morphological features include downslanted palpebral fissures, high narrow palate, dental crowding, and microretrognathia. Ohdo syndrome is responsible for intellectual disability with hyperactivity and aggressive behavior, blepharophimosis, microretrognathia, constipation, and feeding difficulties.30
Recently, 13 individuals were described harboring de novo missense and nonsense MED13 (OMIM 603808) variants. All individuals had mild to moderate intellectual disability and speech disorder. Other features mainly included autism spectrum disorder, attention deficit hyperactivity, optic nerve abnormalities, Duane anomaly, hypotonia, mild congenital heart malformations, and dysmorphism.15 Missense and nonsense MED13L (OMIM 608771) variants are also responsible for moderate to severe intellectual disability and severe speech disorder with poor language. More than 60 individuals have been reported so far. Other features included hypotonia, autism and behavioral disorder, corpus callosum hypoplasia or agenesis, and miscellaneous malformations.14,33,34,35 Finally, disruption of CDK19 has been reported in an individual with intellectual disability, microcephaly, and bilateral congenital retinal folds.13
Therefore, all the subunits of the kinase module seem to be crucial for neurological development. Some features are common between the four conditions linked to MED12, MED13, MED12L, and MED13L such as intellectual disability, behavior abnormalities, and autism spectrum disorders. A CDK19 variant has been reported in only one individual with a different neurological phenotype. Table 2 summarizes common features between the four main MED-related conditions. Corpus callosum anomalies seem common with MED12 andMED12L variants and can be seen less frequently with MED13L variants. MED12 and MED13L variants appear to lead to a more severe condition, with poor speech and facial hypotonia, than MED13 and MED12L related disorders. Finally, the wide range of other organs affected is an inconsistent feature and can be related to the ubiquitous expression of those genes and their broad regulatory role. Mild facial morphological features are common for all those disorders, but do not help in pointing to a clinical diagnosis.
Herein, we demonstrate that MED12L fibroblasts for individuals 1 and 2 show a defect in transcriptional activity as measured by recovery of RNA synthesis after UV irradiation. This defect is similar to the defect observed in a MED12 cell line and milder than the defect seen in a MED13L cell line derived from an individual who showed a more severe neurological phenotype. It can be hypothesized that the pathogenic variants in Mediator kinase module subunits could impair the transcriptional coactivation functions of the complex under specific conditions, during development and during postnatal life, leading to the phenotype observed in the individuals. It has already been similarly postulated that neurodevelopmental defects in individuals affected with CS36,37 and trichothiodystrophy38 could be due to transcriptional defects mediated by variants in the other transcriptional coactivation complexes TFIIH or TFIIE.39
In conclusion, we report here the involvement of MED12L in human disease as has been seen for other subunits of the kinase module of the mediator complex, through transcriptional defect. Even if many intellectual disability syndromes are challenging to differentiate based on clinical phenotype alone, a common clinical pattern seems to emerge for the disorders related to transcriptional defect linked to Mediator complex.
Accession numbers
The accession numbers for the SNPs and CNVs reported in this paper are DECIPHER: 284908, 280845, and 277489; and ClinVar: SCV000611598, SCV000853261, SCV000853262, and SCV000853263.
Change history
03 July 2019
In the Acknowledgements section of the paper the authors neglected to mention that the study was supported by a grant from the National Human Genome Research Institute (NHGRI) UM1HG007301 (SMH, MLT). In addition, the award of MD was associated with the authors Michelle Thompson and Susan Hiatt instead of PhD. The PDF and HTML versions of the Article have been modified accordingly.
References
Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem Sci. 2005;30:235–239.
Conaway RC, Conaway JW. Origins and activity of the Mediator complex. Semin Cell Dev Biol. 2011;22:729–734.
Allen BL, Taatjes DJ. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16:155–166.
Berk AJ. Yin and yang of mediator function revealed by human mutants. Proc Natl Acad Sci USA 2012;109:19519–19520.
Jeronimo C, Robert F. The Mediator complex: at the nexus of RNA polymerase II transcription. Trends Cell Biol. 2017;27:765–783.
Risheg H, Graham JM, Clark RD, et al. A recurrent mutation in MED12 leading to R961W causes Opitz–Kaveggia syndrome. Nat Genet. 2007;39:451–453.
Rump P, Niessen RC, Verbruggen KT, et al. A novel mutation in MED12 causes FG syndrome (Opitz–Kaveggia syndrome). Clin Genet. 2011;79:183–188.
Graham JM, Schwartz CE. MED12 related disorders. Am J Med Genet A. 2013;161A:2734–2740.
Schwartz CE, Tarpey PS, Lubs HA, et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J Med Genet. 2007;44:472–477.
Vulto-van Silfhout AT, de Vries BBA, van Bon BWM, et al. Mutations in MED12 cause X-linked Ohdo syndrome. Am J Hum Genet. 2013;92:401–406.
Isidor B, Lefebvre T, Le Vaillant C, et al. Blepharophimosis, short humeri, developmental delay and Hirschsprung disease: expanding the phenotypic spectrum of MED12 mutations. Am J Med Genet A. 2014;164A:1821–1825.
Donnio L-M, Bidon B, Hashimoto S, et al. MED12-related XLID disorders are dose-dependent of immediate early genes (IEGs) expression. Hum Mol Genet. 2017;26:2062–2075.
Mukhopadhyay A, Kramer JM, Merkx G, et al. CDK19 is disrupted in a female patient with bilateral congenital retinal folds, microcephaly and mild mental retardation. Hum Genet. 2010;128:281–291.
Smol T, Petit F, Piton A, et al. MED13L-related intellectual disability: involvement of missense variants and delineation of the phenotype. Neurogenetics. 2018;19:93–103.
Snijders Blok L, Hiatt SM, Bowling KM, et al. De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder. Hum Genet. 2018;13:675–685.
Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84:524–533.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–930.
Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312: 1870–1879.
Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, Cochran JN, Brothers KB, East KM, Gray DE, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9:43.
Calmels N, Greff G, Obringer C, Kempf N, Gasnier C, Tarabeux J, Miguet M, Baujat G, Bessis D, Bretones P, et al. Uncommon nucleotide excision repair phenotypes revealed by targeted high-throughput sequencing. Orphanet J Rare Dis. 2016;11:26.
Limsirichaikul S, Niimi A, Fawcett H, Lehmann A, Yamashita S, Ogi T. A rapid nonradioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU). Nucleic Acids Res. 2009;37:e31.
Epanchintsev A, Costanzo F, Rauschendorf M-A, Caputo M, Ye T, Donnio L-M, Proietti-de-Santis L, Coin F, Laugel V, Egly J-M. Cockayne’s syndrome A and B proteins regulate transcription arrest after genotoxic stress by promoting ATF3 degradation. Mol Cell. 2017;68:1054–.e6.
Olsen L, Sparsø T, Weinsheimer SM, Dos Santos MBQ, Mazin W, Rosengren A, Sanchez XC, Hoeffding LK, Schmock H, Baekvad-Hansen M, et al. Prevalence of rearrangements in the 22q11.2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: a case-cohort study. Lancet Psychiatry. 2018;5:573–580.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Cai D-C, Fonteijn H, Guadalupe T, Zwiers M, Wittfeld K, Teumer A, Hoogman M, Arias-Vásquez A, Yang Y, Buitelaar J, et al. A genome-wide search for quantitative trait loci affecting the cortical surface area and thickness of Heschl’s gyrus. Genes Brain Behav. 2014;13:675–685.
Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, et al. Panorama of ancient metazoan macromolecular complexes. Nature. 2015;525:339–344.
Martin C-A, Murray JE, Carroll P, Leitch A, Mackenzie KJ, Halachev M, Fetit AE, Keith C, Bicknell LS, Fluteau A, et al. Mutations in genes encoding condensin complex proteins cause microcephaly through decatenation failure at mitosis. Genes Dev. 2016;30:2158–2172.
Clark AD, Oldenbroek M, Boyer TG. Mediator kinase module and human tumorigenesis. Crit Rev Biochem Mol Biol. 2015;50:393–426.
Jeronimo C, Langelier M-F, Bataille AR, Pascal JM, Pugh BF, Robert F. Tail and kinase modules differently regulate core Mediator recruitment and function in vivo. Mol Cell. 2016;64:455–466.
Huang Y, Li W, Yao X, Lin Q-J, Yin J-W, Liang Y, Heiner M, Tian B, Hui J, Wang G. Mediator complex regulates alternative mRNA processing via the MED23 subunit. Mol Cell. 2012;45:459–469.
Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, Shiekhattar R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501.
Graham JM, Visootsak J, Dykens E, Huddleston L, Clark RD, Jones KL, Moeschler JB, Opitz JM, Morford J, Simensen R, et al. Behavior of 10 patients with FG syndrome (Opitz–Kaveggia syndrome) and the p.R961W mutation in the MED12 gene. Am J Med Genet A. 2008;146A:3011–3017.
Muncke N, Jung C, Rüdiger H, Ulmer H, Roeth R, Hubert A, Goldmuntz E, Driscoll D, Goodship J, Schön K, et al. Missense mutations and gene interruption in PROSIT240, a novel TRAP240-like gene, in patients with congenital heart defect (transposition of the great arteries). Circulation. 2003;108:2843–2850.
Asadollahi R, Oneda B, Sheth F, Azzarello-Burri S, Baldinger R, Joset P, Latal B, Knirsch W, Desai S, Baumer A, et al. Dosage changes of MED13L further delineate its role in congenital heart defects and intellectual disability. Eur J Hum Genet. 2013;21:1100–1104.
Adegbola A, Musante L, Callewaert B, Maciel P, Hu H, Isidor B, Picker-Minh S, Le Caignec C, Delle Chiaie B, Vanakker O, et al. Redefining the MED13L syndrome. Eur J Hum Genet. 2015;23:1308–1317.
Kristensen U, Epanchintsev A, Rauschendorf M-A, Laugel V, Stevnsner T, Bohr VA, Coin F, Egly J-M. Regulatory interplay of Cockayne syndrome B ATPase and stress-response gene ATF3 following genotoxic stress. Proc Natl Acad Sci USA 2013;110:E2261–2270.
Wang Y, Chakravarty P, Ranes M, Kelly G, Brooks PJ, Neilan E, Stewart A, Schiavo G, Svejstrup JQ. Dysregulation of gene expression as a cause of Cockayne syndrome neurological disease. Proc Natl Acad Sci USA. 2014;111:14454–14459.
Compe E, Malerba M, Soler L, Marescaux J, Borrelli E, Egly J-M. Neurological defects in trichothiodystrophy reveal a coactivator function of TFIIH. Nat Neurosci. 2007;10:1414–1422.
Kuschal C, Botta E, Orioli D, Digiovanna JJ, Seneca S, Keymolen K, Tamura D, Heller E, Khan SG, Caligiuri G, et al. GTF2E2 mutations destabilize the general transcription factor complex TFIIE in individuals with DNA repair-proficient trichothiodystrophy. Am J Hum Genet. 2016;98:627–642.
Acknowledgements
We thank the patients and their families for participating in this study. The study was supported by a grant from the National Human Genome Research Institute (NHGRI) UM1HG007301 (SMH, MLT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics. M.T.C. and K.M. are employees of GeneDx, Inc., a wholly owned subsidiary of OPKO Health, Inc. The other authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Nizon, M., Laugel, V., Flanigan, K.M. et al. Variants in MED12L, encoding a subunit of the mediator kinase module, are responsible for intellectual disability associated with transcriptional defect. Genet Med 21, 2713–2722 (2019). https://doi.org/10.1038/s41436-019-0557-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0557-3
Keywords
This article is cited by
-
Genome-wide DNA methylation meta-analysis in the brains of suicide completers
Translational Psychiatry (2020)
