
Abstract
The detection of acquired copy-number abnormalities (CNAs) and copy-neutral loss of heterozygosity (CN-LOH) in neoplastic disorders by chromosomal microarray analysis (CMA) has significantly increased over the past few years with respect to both the number of laboratories utilizing this technology and the broader number of tumor types being assayed. This highlights the importance of standardizing the interpretation and reporting of acquired variants among laboratories. To address this need, a clinical laboratory-focused workgroup was established to draft recommendations for the interpretation and reporting of acquired CNAs and CN-LOH in neoplastic disorders. This project is a collaboration between the American College of Medical Genetics and Genomics (ACMG) and the Cancer Genomics Consortium (CGC). The recommendations put forth by the workgroup are based on literature review, empirical data, and expert consensus of the workgroup members. A four-tier evidence-based categorization system for acquired CNAs and CN-LOH was developed, which is based on the level of available evidence regarding their diagnostic, prognostic, and therapeutic relevance: tier 1, variants with strong clinical significance; tier 2, variants with some clinical significance; tier 3, clonal variants with no documented neoplastic disease association; and tier 4, benign or likely benign variants. These recommendations also provide a list of standardized definitions of terms used in the reporting of CMA findings, as well as a framework for the clinical reporting of acquired CNAs and CN-LOH, and recommendations for how to deal with suspected clinically significant germline variants.
Similar content being viewed by others


INTRODUCTION
Genomic testing of hematologic malignancies and solid tumors at the time of disease presentation provides information that is crucial for diagnosis and management. This evaluation may include G-banded chromosome analysis, fluorescence in situ hybridization (FISH) analysis, chromosomal microarray analysis (CMA), gene expression and fusion studies, targeted gene sequencing, as well as gene sequencing panels.
The somatic genomic variants detected in the tumor tissue play a critical role in the patient’s clinical management by aiding in the diagnosis, providing prognostic information, and helping in the choice of appropriate therapy. The types of somatic variants observed include numerical and structural chromosomal abnormalities, single-nucleotide variants (SNVs), nucleotide-level deletions, duplications and insertions (i.e., indels), and gene-level deletions and duplications. One type of somatic structural chromosomal rearrangements common in neoplastic disorders are copy-number abnormalities (CNAs), which are changes that represent acquired gains and losses of chromosomal material. High-resolution genome-wide CMA is being widely used in clinical laboratories to detect acquired CNAs and copy-neutral loss of heterozygosity (CN-LOH) in neoplastic disorders, and is providing important insights into the unique genomic copy-number profile of different tumor tissues. In recent years, the clinical utility of CMA has been well established in the diagnosis of several neoplastic disorders.1,2,3,4,5,6,7
The American College of Medical Genetics and Genomics (ACMG) technical standards and guidelines for CMA in neoplastic disorders includes a section on CMA results evaluation and interpretation.8 This document provided the initial framework for interpreting CMA results in neoplastic disorders; however, it describes broad principles. The use of CMA in neoplastic disorders has significantly increased over the past few years with respect to both the number of laboratories utilizing this technology and the broader number of tumor types being assayed. In practice, the interpretation of CMA results remains relatively subjective and lacks standardization, resulting in inconsistent practices between clinical laboratories. The CNAs and CN-LOH detected by CMA in neoplastic disorders are in many cases critical for optimal patient care. This necessitates standardized interpretation and reporting of acquired variants using an evidence-based system to accurately establish their clinical significance.
To address this need, a clinical laboratory-focused workgroup was established to draft recommendations for the interpretations and reporting of acquired CNAs and CN-LOH in neoplastic disorders. This project is a collaboration between the ACMG Laboratory Quality Assurance (QA) Committee and the Cancer Genomics Consortium (CGC). The workgroup developed recommendations for categorizing CNAs and CN-LOH detected by CMA in neoplastic disorders into specific standard categories (i.e., tiers) of clinical significance based on objective criteria using an evidence-based weighting system. The term “acquired variants” is used in this document to refer to both acquired CNAs and CN-LOH.
These newly developed recommendations, which are described in detail below, include:
- 1.
Standardized definitions of terms used to describe single variants or patterns of variants detected by CMA
- 2.
A four-tier evidence-based categorization system for acquired CNAs and CN-LOH, which is based on the level of available evidence regarding their diagnostic, prognostic, and therapeutic relevance
- 3.
CNA and CN-LOH examples in tiers 1 and 2 in various hematologic malignancies and solid tumors
- 4.
Considerations regarding the interpretation and reporting of unanticipated clinically significant germline variants
- 5.
A framework to standardize the clinical reporting of acquired CNAs and CN-LOH
Even though these proposed technical laboratory standards are intended for interpretation and reporting of acquired variants detected by CMA in neoplastic disorders, the newly developed recommendations should be applicable to acquired structural variants (including CNAs) detected by sequencing-based approaches, as the clinical testing practices move increasingly toward these technologies.
METHODS
These technical laboratory standards were informed by a review of the literature and current guidelines. Resources consulted included PubMed; current World Health Organization (WHO) and National Comprehensive Cancer Network (NCCN) guidelines; and relevant ACMG, Association for Molecular Pathology (AMP), and College of American Pathologists (CAP) guidelines. The workgroup members also used their expert opinion and empirical data to inform their recommendations. The proposed four-tier evidence-based categorization system for CNAs and CN-LOH was refined and extensively tested among the workgroup members using clinical cases from the members’ diagnostic laboratories. In addition, input from the greater cancer genomics community was solicited during the annual CGC meeting when this tier system was first presented. Any conflicts of interests for workgroup members are listed at the end of the paper. The ACMG Laboratory QA Committee reviewed the document providing further input on the content, and a final draft was presented to the ACMG Board of Directors for review and approval to post on the ACMG website for member comment. Upon posting to the ACMG website, an email and link were sent to all ACMG members inviting participation in the 30-day open comment process. All members’ comments and additional evidence received were assessed by the authors, and these recommendations were incorporated into the document as deemed appropriate. Member comments and author responses were reviewed by representatives of the ACMG Laboratory QA Committee and the ACMG Board of Directors. The final document was approved for publication by the ACMG and the CGC Board of Directors.
Definitions of specific terms used to describe single variants or patterns of variants detected by CMA
Proposed four-tier evidence-based categorization system for acquired CNAs and CN-LOH
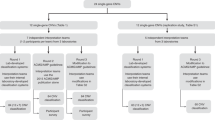
The interpretation of clinical significance of acquired genomic variants is based on their impact on clinical care, including diagnostic, prognostic, and therapeutic significance. The weight of clinical impact of a genomic variant is gauged by the level of available evidence regarding its association with a specific diagnosis, disease outcome, and/or response to a particular treatment. The evidence used for variant categorization is weighted differently based on its likely impact on clinical decision-making. Additionally, the type and size of study providing this evidence is considered in the interpretation of the variants, with professional practice guidelines, large collaborative studies, and replicated studies carrying more weight than individual case reports. Based on literature review and workgroup consensus, the workgroup proposes categorizing genomic variants detected by CMA in neoplastic disorders into four tiers according to the level of evidence for their clinical significance as described below (Fig. 1). The workgroup adapted the levels of evidence published by the Oxford Centre for Evidence-Based Medicine (CEBM).12 An attempt was made to keep these newly developed recommendations aligned, to the extent possible, with the recently published standards and guidelines for the interpretation and reporting of sequence variants in cancer.13 The sequence variant guidelines introduce the concept that the interpretation of somatic variants should focus on their impact on clinical care. In addition to their oncogenic role, they may be associated with a favorable or adverse prognosis, with sensitivity, resistance, or toxicity to a specific therapy, with eligibility for clinical trials, and/or with better diagnostic accuracy. The principles put forward for interpretation of somatic sequence variants are applicable for interpretation of somatic CNAs and CN-LOH, which can also serve as biomarkers of prognosis, sensitivity, or resistance to targeted therapies, and/or can support a diagnosis of a particular tumor type. In addition, there is an increasing trend in genomic oncology testing to use consolidated sequencing-based assays to detect somatic SNVs, indels, CNAs, and abnormal gene fusions in selected cancer-related genes. Reporting results of such integrated assays would not be practical if disparate sets of rules had to be applied for interpretation of SNVs and indels versus CNAs and CN-LOH. With the prediction that unbiased genome-wide evaluation for different types of genetic and genomic variants (including both sequence variants and numerical and structural chromosome rearrangements) may become feasible for cancer samples in the near future, a unified approach for the clinical interpretation, classification, and reporting of all somatic variants will become a necessity.

Four-tier evidence-based categorization system for acquired copy-number abnormalities (CNAs) and copy-neutral loss of heterozygosity (CN-LOH) detected by chromosomal microarray analysis (CMA).CEBM Oxford Centre for Evidence-Based Medicine, COG Children’s Oncology Group, COSMIC Catalogue of Somatic Mutations in Cancer, IPSS International Prognostic Scoring System for myelodysplastic syndromes, NCCN National Comprehensive Cancer Network, WHO World Health Organization.
Tables 1 and 2 provide examples of CNAs and CN-LOH in tiers 1 and 2 in various hematologic malignancies and solid tumors.
- I.
Tier 1 (variants with strong clinical significance): Variants with strong diagnostic, prognostic, and/or therapeutic clinical significance. They have been demonstrated to play a critical role in the oncogenic process under investigation. Based on the level of evidence available, tier 1 variants are further subdivided into:
- a.
Tier 1A: Acquired variants or a specific pattern of acquired variants that fulfill one or more of the following criteria:
- –
Define a specific entity in the WHO classification.
- –
Are included in professional clinical practice guidelines as clinically significant variants (e.g., NCCN, Children’s Oncology Group (COG), Myelodysplastic Syndromes (MDS) International Prognostic Scoring System, International Myeloma Working Group Criteria).
- –
Can be treated by a targeted FDA approved drug.
Tier 1A also includes germline pathogenic variants associated with cancer predisposition.
- –
- b.
Tier 1B: Acquired variants or a specific pattern of acquired variants with either:
- –
High quality evidence (levels 1 and 2 CEBM evidence) in the literature that shows association with a specific neoplasm, prognosis, or treatment response. This includes well-powered studies in the form of randomized controlled clinical trials, systematic review and meta-analysis of these studies, and cohort studies with consensus from experts in the field.
- –
Good quality evidence (level 3 CEBM evidence) in the literature that shows association with a specific neoplasm, prognosis, or treatment response. This includes multiple (at least two) smaller clinical studies in the form of cohort or case–control studies that have been confirmed and reproduced by different independent groups.
- –
- a.
- II.
Tier 2 (variants with some clinical significance): Acquired variants or a specific pattern of acquired variants with some diagnostic, prognostic, and/or therapeutic clinical significance. They include:
- –
Recurrent variants observed in different neoplasms but not specific to a particular tumor type, and usually encompassing Catalogue of Somatic Mutations in Cancer (COSMIC) census cancer genes(s).
- –
Acquired variants or a specific pattern of acquired variants with average quality evidence (levels 4 and 5 CEBM evidence) in the literature that shows association with a specific neoplasm, prognosis, or treatment response. This includes a small case series or multiple (at least two) case reports that describe the association.
- –
- III.
Tier 3 (clonal variants with no documented neoplastic disorder association): Acquired clonal variants with no documented neoplastic disorder association. All variants that do not meet the criteria for tiers 1 and 2 and cannot be classified as constitutional benign or likely benign, can be classified as tier 3 variants. Tier 3 variants are defined as “acquired clonal variants with no documented neoplastic disorder association” rather than “acquired clonal variants with uncertain clinical significance.” This is because an “acquired clonal variant” is by default significant for this particular patient because it can be used as a marker for the neoplastic clone to monitor residual disease and/or relapse.
- IV.
Tier 4 (benign or likely benign variants): Constitutional benign or likely benign variants that are listed in the ClinGen curated benign variants and/or in the Database of Genomic Variants (DGV) with ≥1% population frequency, and usually do not encompass COSMIC cancer gene(s). It is not recommended to report tier 4 variants.
General and special considerations
-
1.
The interpretation of clinical significance of CNAs and CN-LOH using this tier system should be performed in the context of the clinical/pathologic diagnosis, as well as other laboratory tests including G-banded karyotype, FISH, and other relevant tests. This is crucial because some acquired variants will have different clinical significance in different neoplastic disorders. For example, 1q gain is associated with adverse prognosis in multiple myeloma (MM) (tier 1A),14,15 while it does not have major prognostic significance in MDS (tier 2).16 CNAs may also have different clinical significance depending on other cytogenetic or molecular diagnostic abnormalities present in the tumor. For example, loss of chromosome 7 or 7q deletion are typically associated with an inferior outcome in myeloid malignancies (tier 1A), but in acute myeloid leukemia (AML) with aCBFB gene rearrangement, they do not appear to significantly change the prognosis (tier 2).17
-
2.
This tier system can be used to classify a specific pattern of CNAs and/or CN-LOH that is diagnostic of a specific neoplastic disease entity. This includes a characteristic pattern of whole chromosome gains/losses (e.g., hyperdiploid and hypodiploid B-ALL) and whole chromosome CN-LOH (e.g., doubled hypodiploid/near-haploid B-ALL).18 It also includes a characteristic signature of gains and losses along one chromosome (e.g., intrachromosomal amplification of chromosome 21 [iAMP21] in B-ALL).19 The pattern of acquired gains/losses can be classified collectively using the tier system.
-
3.
Diagnostic balanced chromosomal abnormalities (e.g., translocations, inversions, and insertions) detected by G-banded karyotype and/or FISH testing but not by CMA should be discussed in the CMA report but should not be included in the classification using the tier system or listed in the results table/nomenclature string. When present in the unbalanced form and detected by CMA with breakpoints mapping within genes known to be associated with a specific gene fusion, these abnormalities can be classified using the tier system and listed in the results table/nomenclature string (e.g., the presence of an extra copy of the Philadelphia chromosome der(22)t(9;22)(q34;q11.2) in CML or ALL,18 or an extra copy of the der(21)t(12;21)(p13;q22) in B-ALL, and the unbalanced der(19)t(1;19)(q23;p13) in B-ALL).20
-
4.
An interstitial loss or gain involving one chromosome arm with recurring breakpoints in genes known to be involved in a specific gene fusion can be classified using this tier system (e.g., 4q12 deletion that results in FIP1L1-PDGFRA fusion, PAR1 deletion at Xp22.33/Yp11.32 that results in P2RY8-CRLF2 fusion, and 9q34.1 gain that results inNUP214-ABL1 fusion).18,21
-
5.
Interstitial or terminal losses or gains involving two chromosome arms with breakpoints within genes known to be associated with a specific gene fusion as a result of an interchromosomal rearrangement (e.g., translocation or insertion) or intrachromosomal rearrangement (e.g., inversion) should be interpreted according to the level of supporting evidence. They can be classified using this tier system with later confirmation of the gene fusion by other molecular techniques if there is enough supporting evidence, including the clinical/pathologic diagnosis, visible recurrent rearrangement by G-banded karyotype, and/or other acquired variant known to be associated with the gene fusion in question. In the absence of such supporting evidence, the report should describe the possibility of a gene fusion but without classifying the variants using the tier system until the fusion is confirmed by other molecular techniques.
-
6.
Correlation of the CMA results with the G-banded karyotype and FISH results is strongly recommended because some professional clinical practice guidelines used to classify tier 1A variants are technique specific. For example, some chromosomal abnormalities can only be considered diagnostic/prognostic if detected by G-banded karyotype (e.g., MDS and MM prognostic criteria).
-
7.
CMA has the potential to identify acquired variants associated with comorbid neoplastic disorders. For example, comorbid MDS-related variants may be identified in patients treated for chronic lymphocytic leukemia (CLL) or MM either because of prior therapy or age-related disease. These variants should be interpreted in the context of the clinical/pathologic diagnosis and correlated with G-banded karyotypes from both stimulated and unstimulated CLL or MM cultures. CMA performed in MM on CD138+ enriched cells is helpful in identifying MM-specific acquired variants.7
-
8.
The term “CN-LOH” is used in this document to refer to a region with acquired allelic imbalance (i.e., homozygosity) without an associated copy-number change (i.e., copy-neutral), which is a common finding in cancer. The term “copy-neutral” is used to allow distinction from loss of heterozygous single-nucleotide polymorphism (SNP) calls due to a one copy-number loss (i.e., heterozygous deletion). However, in some cases LOH can also be observed with a copy-number gain. Examples include high-level amplification involving only one allele, and the copresence of a clone with trisomy of a particular chromosome and a subclone that lost one copy of that chromosome resulting in whole chromosome LOH.
-
9.
Regions of CN-LOH may have a higher level of clinical significance if they span a gain-of-function variant in an oncogene and/or loss-of-function variant in a tumor suppressor gene documented in this patient. This is especially relevant in laboratories that do integrated reporting of CNAs, regions of CN-LOH, and sequence variants results.
Databases and resources for interpretation of CNAs and CN-LOH in neoplastic disorders
A wealth of genomic information has been generated for different tumor types through chromosome analysis and large-scale genome sequencing projects, and the data have been consolidated into many public databases. However, the majority of such databases house information at a gene and variant level, and resources focused on incidence and significance of acquired CNAs and CN-LOH in neoplastic disorders are limited. In the absence of CNA-specific information, gene and variant-centered databases can be used to support interpretation of CNAs involving specific genes.
To allow utilization of gene and variant-focused data for informing interpretation of CNAs and CN-LOH in oncology samples, it is important to annotate the mechanism of action for genes and variants related to cancer. Such mechanisms typically include loss of function of tumor suppressors, gain of function of oncogenes, abnormal gene fusions, and translocations involving regulatory regions. If variants affecting a gene are proven to be loss-of-function variants, it can be extrapolated that a deletion of the same gene or a larger region containing that gene would also confer a loss of function.
A brief overview of resources that are useful in interpretation of CMA results in oncology is provided in Table 3. Such resources include:
- 1.
Databases and data portals focusing directly on acquired CNAs and CN-LOH
- 2.
Databases and data portals focusing on acquired sequence variants, which allow the evaluation of whether specific genes within the region affected by a CNA have been associated with the tumor type of interest
- 3.
Knowledge bases that contain curated information on the significance of individual genes and acquired sequence variants in different tumor types
- 4.
Chromosome-level databases and knowledge bases that compile data from conventional cytogenetic analysis and curations regarding the significance of chromosome aberrations detected by karyotyping
- 5.
Databases of benign and pathogenic germline variants that allow exclusion of benign germline variants and interpretation of germline secondary findings
To facilitate review and interpretation of acquired CNAs data, laboratories are advised to curate and maintain lists of genes and regions of clinical relevance in a variety of tumor types. These lists support comprehensive and efficient recognition of disease-relevant loci, and allow consistency in interpretation. A laboratory can also opt to develop lists of predefined pertinent positives and negatives per tumor type, and perform systematic careful evaluation for their presence.
The curated clinical-grade disease-specific gene lists can be maintained in both a spreadsheet and .bed file format. It is useful for such lists to be converted into a format compatible with the CMA software, so they can be uploaded and used as custom annotation tracks during case review. This allows quickly recognizing acquired CNAs that contain genes implicated in the tumor of interest. A comprehensive list of genes shown to contain variants causally implicated in cancer (so-called Cancer Gene Census) is maintained in the COSMIC database, and can be downloaded from its website (https://cancer.sanger.ac.uk/census).
Because the databases and data portals for search and visualization of acquired CNAs in neoplastic disorders are rare, clinical interpretation typically requires a review of primary literature. Such interpretation remains a complex and time-consuming task that requires appropriate professional training and certification in clinical cytogenetics and/or molecular diagnostics. It also necessitates familiarity with the CMA assay and an understanding of the specific tumor biology.
Considerations regarding interpretation and reporting of unanticipated clinically significant germline variants
In addition to acquired clonal abnormalities, genome-wide analysis of tumor samples also detects constitutional germline copy-number variants (CNVs) and regions of absence of heterozygosity (AOH). These may include benign population variants, germline CNVs directly related to the neoplasm under investigation (e.g., germline deletions of tumor suppressor genes), and pathogenic CNVs that are diagnostic or predictive of a presymptomatic or unrecognized genetic condition unrelated to the patient’s tumor. With the exception of CNVs that are associated with an increased risk of neoplasia, other germline variants are unanticipated and unrelated to the reason for CMA. Referring clinicians must have a clear understanding of the potential for these discoveries; the best practice would also include informing the patients and their families about the possibility of secondary findings before the test is ordered, and implementing a formal informed consent process. Before offering clinical CMA testing for oncology samples, laboratories should develop a process for appropriate follow-up if an unanticipated, likely germline abnormality is observed.
Indications that a detected CNV or AOH may be germline
Distinguishing between acquired CNAs/CN-LOH and constitutional CNVs/AOH in CMA may be challenging. The possibility that a variant may be germline should be considered in the following scenarios:
- 1.
Involvement of 100% of the cells in a sample. Often, acquired variants involve only a subset of cells corresponding to the tumor clone. Review of the log2 ratio and SNP data allows determining if a variant is present in all or only a subset of cells in a sample. However, it is important to be aware of the following caveats:
- a.
Some specimens may consist of pure tumor tissue and have acquired variants that involve close to 100% of the cells (e.g., a bone marrow specimen packed with leukemic blasts or a dissected tumor section).
- b.
Copy-number losses encompassing cancer predisposition genes are particularly difficult to interpret by CMA alone. For small abnormalities with insufficient SNP data, CMA may not reliably differentiate a heterozygous loss in 100% of the cells from a homozygous loss in 50% of the cells. If CMA shows copy-number losses encompassing cancer predisposition genes, it might not be possible to distinguish between a germline heterozygous deletion of the gene in question in 100% of the cells versus acquired biallelic loss of the gene in 50% of the cells. Frequently encountered examples include the Fanconi anemia/DNA repair pathway genes (includingBRCA1 andBRCA2),NF1, RB1, and PAX5. Follow-up interphase FISH analysis using gene-specific probes can be helpful in distinguishing between these two possibilities.
- a.
- 2.
Higher proportion of cells involved by a variant than expected by pathologic findings. For hematologic malignancies, a finding may be germline if it appears to involve a significantly greater proportion of cells than that expected based on the blast cell count or degree of involvement determined by morphology or flow cytometry. Correlation with hematopathology and flow cytometry/immunophenotyping data is valuable, and efforts to obtain this information are recommended. For solid tumors, a finding may be germline if the estimate of involvement by CMA is significantly greater than the estimation of tumor cell fraction provided by the submitting pathologist. However, estimating tumor fraction in solid tumors is often challenging and involves subjective judgment; this estimate may not always be perfectly correlated with CMA results.
- 3.
Supporting clinical information may suggest that a CMA variant is germline:
- a.
Some tumor types are frequently associated with the presence of predisposing germline variants. Examples include Wilms tumor, tuberous sclerosis complex (TSC1/TSC2) tumors, neurofibromas, adrenocortical carcinoma, and rhabdoid tumor (Supplementary Table 1). Laboratories should have an increased level of suspicion for germline variants when performing CMA for these tumor types.
- b.
CNVs/AOH including known cancer predisposition genes (Supplementary Table 1) may be suspected as germline in patients with features of hereditary cancer syndromes, including diagnosis at unusually young age, development of bilateral or multifocal tumors, or family/personal history of cancer.
- c.
CNVs involving genes and regions associated with known pathogenic microdeletion/microduplication syndromes may be suspected as germline in patients who have reported features consistent with the disorder in question. If the provided clinical information is limited, the laboratory may request additional details to allow accurate interpretation of the findings.
- a.
Interpretation and reporting of suspected germline variants
When reporting variants that are suspected to be germline, CNVs predisposing to cancer should be distinguished from variants unrelated to the patient’s cancer diagnosis.
Germline CNVs directly related to the neoplasm under investigation (e.g., germline deletion of a tumor suppressor gene) should be reported as being of strong clinical significance (tier 1A) and discussed in the interpretation section of the report. This includes germline CNVs involving cancer predisposition genes listed in the 2016 ACMG secondary findings document.22
For likely germline CNVs not related to the neoplasm under investigation:
- a.
Laboratories should have an established policy for reporting CNVs that are likely germline and have been curated as pathogenic by ClinGen (including pathogenic CNVs associated with disorders that show incomplete penetrance) and/or span known haploinsufficient or triplosensitive genes.23 These findings can influence clinical care for the patient and the family; as such, they should be included in the report and discussed as potentially constitutional clinically significant variants (see below).
- b.
Possibly constitutional CNVs unrelated to the patient’s cancer diagnosis should not be classified into the tier system. For unambiguous reporting, the laboratory may have a separate section of the report for describing these variants.
Follow-up recommendations for suspected germline variants
If CMA of a tumor sample detects suspected germline CNVs/AOH, the report should contain recommendations for appropriate follow-up, including the following:
- a.
Referral to a genetic specialist for evaluation and counseling.
- b.
Confirmation of germline status by testing noninvolved tissue. For patients with solid tumors, a peripheral blood sample may be tested. For patients with hematologic malignancies, the optimal samples for germline testing are cultured skin fibroblasts, although a buccal swab or a peripheral blood sample at the time of complete remission may be acceptable.
Suggested language for reporting suspected constitutional findings:
Suspected germline variant
Based on (percent of cells involved, supporting clinical information, etc.), this finding may represent a germline variant. Genetic testing of a tissue that is not involved in the neoplastic process is recommended when the patient is in clinical remission to determine whether this is a germline or an acquired variant and to aid in determination of its clinical significance. If the variant is germline, genetic counseling is recommended for additional information about this variant and its clinical significance.
Reporting recommendations for acquired CNAs and CN-LOH
The laboratory must ensure that the clinical report accurately describes the findings and clearly communicates their clinical significance. The report should include the preanalytic, analytic, and postanalytic factors that are relevant to the clinical interpretation of the findings, as well as elements that represent regulatory requirements (which are outlined in the ACMG Laboratory Standards and Guidelines; Section E8). Despite the large amount of information that must be included, the report should be as simple and concise as possible, formatted in a way that allows the results to be easily seen and understood, and the clinically critical information should appear at the beginning. Displaying the results in tables may be helpful to increase the overall clarity of the report, provided that the tables can be integrated into the medical record.
In contrast to reporting results of CMA testing for constitutional variants, reports for oncology specimens should not be limited to positive findings. In some cases, what the test does not detect may be of the same or even greater significance than the positive findings. It is strongly recommended that pertinent negatives relevant for clinical management are included in a disease-specific manner. This will typically include tier 1A variants that are used for clinical decision-making (as key prognostic markers or predictors of response or resistance to targeted treatments).
Detected CNAs and CN-LOH should be classified into the four-tier system described above. In complex cases, laboratories may opt not to specify tier classification for every variant individually, but should accurately point out and discuss in the interpretation section all the variants with strong or some clinical significance (tiers 1 and 2). It is not recommended that tier 4 variants (benign/likely benign) be included in the report.
If there is doubt about a variant being “acquired/clonal” versus “germline/constitutional,” this should be discussed in the report, and such variants should not be tiered using the classification system for acquired variants until this uncertainty is clarified.
For clear communication of the relevant and required information, it is recommended that the CMA clinical report be organized into the following sections: results, interpretation, recommendations (if applicable), references, and method description and disclaimers.
Results section
CMA results should be reported according to the current version of the International System for Human Cytogenomic Nomenclature (ISCN).24 According to ISCN 2016, results can be reported using a table, as a nomenclature string, or both at the discretion of the laboratory director. If the results are displayed in a table, the following information should be included:
Required
Chromosomes and corresponding bands involved in the variant
Type of variant (loss, gain, amplification, CN-LOH)
Genomic coordinates with designated genome build
Recommended
Copy-number state and percentage of cells involved, estimated based on the log2 ratio and SNP data
Tier classification
Optional
Variant size in kb or Mb
COSMIC cancer census genes within the affected region
Variants that constitute a diagnostic pattern may be classified collectively in the table using the tier system. The results table can be included either at the beginning or at the end of the report. In complex cases, it may be helpful to clinicians to emphasize clinically significant findings at the beginning of the report, and to place the complete results table at the end. In such cases, laboratories should consider including an additional abridged summary table with clinically significant variants at the beginning of the report.
While the clone structure cannot be ascertained with certainty by CMA, it is recommended to report the approximate percentage of cells (levels of mosaicism) for acquired variants to give an estimate of possible clones and subclones.
Full interpretation of clinically significant variants and a text summary integrating results
The full interpretation should include comments on the following variants:
Clinically significant CNAs and/or CN-LOH (tier 1 and 2 variants).
Clinically significant pattern of CNAs and/or CN-LOH (tier 1 and 2 variants).
CNAs and/or CN-LOH of potential clinical significance (cannot be tiered at the time of reporting). This category addresses point 5 in “General and special considerations” when there is uncertainty about an acquired variant being indicative of a specific gene fusion in the absence of supporting evidence at the time of reporting.
Optional: other clonal variants (tier 3 variants).
The comments may contain information about the prevalence and functional, prognostic, or predictive significance of the detected CNAs or CN-LOH in a particular tumor type. The laboratory may want to specifically point out the presence of abnormalities that are associated with response to a targeted treatment, in particular if they predict sensitivity to an FDA approved drug. However, specific treatment recommendations are not encouraged. A text summary should integrate CMA results and correlate them with the results of G-banded karyotype and FISH studies. This summary can be included at the beginning or at the end of the interpretation section. Key abnormalities detected by karyotyping and FISH should not be classified into tiers, but should be discussed in the summary with correlation to the CMA findings.
Recommendations
A recommendation section may be included when necessary based on the findings. For example, appropriate follow-up should be recommended in cases in which CMA findings may be germline (see “Follow-up recommendations for suspected germline variants”). Recommendations should also include molecular confirmation of clinically significant abnormalities that are predicted but cannot be established based solely on CMA results (this includes breakpoints suggestive of a particular abnormal gene fusion, CN-LOH suggestive of a variant in a particular oncogene or a tumor suppressor gene, etc.). Treatment recommendations (for the use of specific targeted therapies or enrollment into specific clinical trials) typically should not be included, considering that a treatment choice depends on many factors (other than the diagnosis provided on a test requisition and the CMA findings) that are unknown to the laboratory.
References
Key publications that were used as evidence to classify detected variants into tiers should be listed in the final report.
Methodology and disclaimers
Methodologic details should be presented at the bottom of the report and should include a brief description of the array platform and assay performance characteristics; this may include size resolution and limitations of the assay (e.g., lack of sensitivity for detecting abnormalities present in a low proportion of cells in the sample, inaccuracy in ploidy determination, inability to detect balanced rearrangements, etc.). Criteria for inclusion of findings in the report and criteria for tier classification should be briefly stated.
The order of different report sections is at the discretion of the laboratory director. Laboratories should have the freedom to choose their own reporting format as long as the report includes the required information outlined above and clearly communicates clinically relevant findings. Laboratory report formats may be limited by a specific reporting system used by the associated hospital, medical center, or commercial entity. Several report examples for different tumor types, including cases with both simple and complex findings, are provided in the supplementary materials of this document.
Summary
The technical standards for interpretation and reporting of acquired CNAs and CN-LOH in neoplastic disorders described were developed in response to an urgent need to standardize the interpretation and reporting of these acquired variants using an evidence-based system with objective criteria. These recommendations represent an expert consensus of the workgroup members based on literature review, empirical data, and their professional judgment. These recommendations describe a four-tier evidence-based categorization system for acquired CNAs and CN-LOH. They outline the variant classification criteria for each tier based on the level of evidence available, and provide examples in tiers 1 and 2 in various hematologic malignancies and solid tumors. This document also provides a list of standardized definitions of terms used in the reporting of these variants, and recommendations for handling suspected clinically significant germline variants. Finally, this document outlines a framework for the clinical reporting of acquired CNAs and CN-LOH. The workgroup believes that the technical standards presented here will help clinical laboratories in achieving better standardized interpretation of CMA results. The workgroup will be constantly reviewing and revising these recommendations based on feedback from the cancer genomic community through a follow-up evaluation mechanism established in collaboration with the ACMG and CGC.
Supplementary materials
-
Supplementary Table 1 illustrating selected tumor suppressor genes associated with germline predisposition to cancer.
-
CMA report examples in hematologic malignancies and solid tumors.
-
Supplementary figures illustrating examples of amplification, chromothripsis, intrachromosomal complexity, and genomic complexity. The same pattern of acquired CNAs suggestive of a specific disease entity is demonstrated using different CMA platforms. To illustrate the clinical utility of this tier classification system in the interpretation of acquired CNAs derived from whole genome sequencing (WGS), examples of such abnormalities derived from WGS data are also included.
References
Dougherty MJ, Tooke LS, Sullivan LM, Hakonarson H, Wainwright LM, Biegel JA. Clinical utilization of high-resolution single nucleotide polymorphism based oligonucleotide arrays in diagnostic studies of pediatric patients with solid tumors. Cancer Genet. 2012;205:42–54.
Roth JJ, Santi M, Rorke-Adams LB, et al. Diagnostic application of high resolution single nucleotide polymorphism array analysis for children with brain tumors. Cancer Genet. 2014;207:111–123.
Wang Y, Miller S, Roulston D, Bixby D, Shao L. Genome-wide single-nucleotide polymorphism array analysis improves prognostication of acute lymphoblastic leukemia/lymphoma. J Mol Diagn. 2016;18:595–603.
Xu X, Bryke C, Sukhanova M, et al. Assessing copy number abnormalities and copy-neutral loss-of-heterozygosity across the genome as best practice in diagnostic evaluation of acute myeloid leukemia: an evidence-based review from the Cancer Genomics Consortium (CGC) myeloid neoplasms working group. Cancer Genet. 2018;228-229:218–235.
Kanagal-Shamanna R, Hodge JC, Tucker T, et al. Assessing copy number aberrations and copy neutral loss of heterozygosity across the genome as best practice: an evidence based review of clinical utility from the Cancer Genomics Consortium (CGC) working group for myelodysplastic syndrome, myelodysplastic/myeloproliferative and myeloproliferative neoplasms. Cancer Genet. 2018;228-229:197–217.
Chun K, Wenger GD, Chaubey A, et al. Assessing copy number aberrations and copy-neutral loss-of-heterozygosity across the genome as best practice: an evidence-based review from the Cancer Genomics Consortium (CGC) working group for chronic lymphocytic leukemia. Cancer Genet. 2018;228-229:236–250.
Pugh TJ, Fink JM, Lu X, et al. Assessing genome-wide copy number aberrations and copy-neutral loss-of-heterozygosity as best practice: an evidence-based review from the Cancer Genomics Consortium working group for plasma cell disorders. Cancer Genet. 2018;228-229:184–196.
Cooley LD, Lebo M, Li MM, Slovak ML, Wolff DJ, Working Group of the American College of Medical Genetics and Genomics (ACMG) Laboratory Quality Assurance Committee. American College of Medical Genetics and Genomics technical standards and guidelines: microarray analysis for chromosome abnormalities in neoplastic disorders. Genet Med. 2013;15:484–494.
Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71.
Hirsch D, Kemmerling R, Davis S, et al. Chromothripsis and focal copy number alterations determine poor outcome in malignant melanoma. Cancer Res. 2013;73:1454–1460.
Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell. 2013;152:1226–1236.
Howick J, Chalmers I, Glasziou P, et al. Oxford Centre for Evidence-Based Medicine (OCEBM) Levels of Evidence Working Group. The 2011 Oxford Levels of Evidence. May 1st 2016. https://www.cebm.net/index.aspx?o=5653.
Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23.
Rajkumar SV. Updated diagnostic criteria and staging system for multiple myeloma. Am Soc Clin Oncol Educ Book. 2016;35:e418–23.
Shah GL, Landau H, Londono D, et al. Gain of chromosome 1q portends worse prognosis in multiple myeloma despite novel agent-based induction regimens and autologous transplantation. Leuk Lymphoma. 2017;58:1823–1831.
Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System (IPSS-R) for myelodysplastic syndromes. Blood. 2012;120:2454–2465.
Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities amongst 5,876 younger adult patients treated in the UK Medical Research Council trials. Blood. 2010;116:354–365.
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405.
Harrison CJ. Blood spotlight on iAMP21 acute lymphoblastic leukemia (ALL), a high-risk pediatric disease. Blood. 2015;125:1383–1386.
Baughn LB, Biegel JA, South ST, et al. Integration of cytogenomic data for furthering the characterization of pediatric B-cell acute lymphoblastic leukemia: a multi-institution, multi-platform microarray study. Cancer Genet. 2015;208:1–18.
KG Roberts, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–1015.
Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SFv2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255.
Riggs ER, Church DM, Hanson K, et al. Towards an evidence-based process for the clinical interpretation of copy number variation. Clin Genet. 2012;81:403–412.
McGowan-Jordan J, Simons A, Schmid M. An international system for human cytogenomic nomenclature. Basel, Switzerland: S Karger; 2016.
Acknowledgements
The authors would like to thank Beth Pitel and other members of the Mayo Clinic Genomics of Oncology Annotation Team (GOAT) for their valuable input regarding resources for interpretation of CNAs and CN-LOH in neoplastic disorders.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
All members of this workgroup are directors of clinical laboratories that use chromosomal microarray technologies.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Disclaimer:
This laboratory standard is designed primarily as an educational resource for clinical laboratory geneticists to help them provide quality clinical laboratory genetic services. Adherence to this standard is voluntary and does not necessarily assure a successful medical outcome. This standard should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, the clinical laboratory geneticist should apply his or her own professional judgment to the specific circumstances presented by the individual patient or specimen.
Clinical laboratory geneticists are encouraged to document in the patient’s record the rationale for the use of a particular procedure or test, whether or not it is in conformance with this standard. They also are advised to take notice of the date any particular standard was adopted, and to consider other relevant medical and scientific information that becomes available after that date. It also would be prudent to consider whether intellectual property interests may restrict the performance of certain tests and other procedures.
The Board of Directors of the American College of Medical Genetics and Genomics approved this technical standard on 25 March 2019. The Board of Directors of the Cancer Genomics Consortium approved this technical standard on 27 March 2019.
Supplementary information
Rights and permissions
About this article

Cite this article
Mikhail, F.M., Biegel, J.A., Cooley, L.D. et al. Technical laboratory standards for interpretation and reporting of acquired copy-number abnormalities and copy-neutral loss of heterozygosity in neoplastic disorders: a joint consensus recommendation from the American College of Medical Genetics and Genomics (ACMG) and the Cancer Genomics Consortium (CGC). Genet Med 21, 1903–1916 (2019). https://doi.org/10.1038/s41436-019-0545-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0545-7
Keywords
This article is cited by
-
CCDC58 is a potential biomarker for diagnosis, prognosis, immunity, and genomic heterogeneity in pan-cancer
Scientific Reports (2024)
-
From the identification of actionable molecular targets to the generation of faithful neuroblastoma patient-derived preclinical models
Journal of Translational Medicine (2024)
-
Detection of MET amplification by droplet digital PCR in peripheral blood samples of non-small cell lung cancer
Journal of Cancer Research and Clinical Oncology (2023)
-
Deriving tumor purity from cancer next generation sequencing data: applications for quantitative ERBB2 (HER2) copy number analysis and germline inference of BRCA1 and BRCA2 mutations
Modern Pathology (2022)
-
Proposal of Diagnostic Approach of Periodontal Primary Non Hodgkin Lymphoma of Bone with Flow Cytometry as an Essential Diagnostic Component
Head and Neck Pathology (2022)
