
Abstract
Purpose
Skeletal muscle growth and regeneration rely on muscle stem cells, called satellite cells. Specific transcription factors, particularly PAX7, are key regulators of the function of these cells. Knockout of this factor in mice leads to poor postnatal survival; however, the consequences of a lack of PAX7 in humans have not been established.
Methods
Here, we study five individuals with myopathy of variable severity from four unrelated consanguineous couples. Exome sequencing identified pathogenic variants in the PAX7 gene. Clinical examination, laboratory tests, and muscle biopsies were performed to characterize the disease.
Results
The disease was characterized by hypotonia, ptosis, muscular atrophy, scoliosis, and mildly dysmorphic facial features. The disease spectrum ranged from mild to severe and appears to be progressive. Muscle biopsies showed the presence of atrophic fibers and fibroadipose tissue replacement, with the absence of myofiber necrosis. A lack of PAX7 expression was associated with satellite cell pool exhaustion; however, the presence of residual myoblasts together with regenerating myofibers suggest that a population of PAX7-independent myogenic cells partially contributes to muscle regeneration.
Conclusion
These findings show that biallelic variants in the master transcription factor PAX7 cause a new type of myopathy that specifically affects satellite cell survival.
Similar content being viewed by others

INTRODUCTION
PAX7 is a member of the paired box gene family, the members of which all share a highly conserved DNA binding domain, the paired box. There are nine different PAX genes, which play important roles in organogenesis and tissue development by regulating the lineage determination and the maintenance of progenitor cells.1 Loss-of-function variants in all of the PAX genes have been associated with human diseases, except for PAX7.2 A chromosomal translocation resulting in the fusion of PAX7 to FOXO1 has been shown to be associated with alveolar rhabdomyosarcoma;3 however, the impact of PAX7 loss-of-function in human tissue development and pathology is still elusive.
The role of Pax7 has been widely investigated in animal studies. The expression pattern points toward a role in the development of cephalic neural crest derivatives, the central nervous system, and skeletal muscle. Pax7-/- mice appear normal during embryonic development but they die shortly after weaning.4 They exhibit malformations in facial structures involving the maxilla and the nose, but no obvious central nervous system or skeletal muscle abnormalities at birth. Neural and skeletal muscle development during mouse embryogenesis relies mainly on Pax3, a paralogue of Pax7 that shares a similar structure (Suppl. Fig. S1) and partially overlapping function, which may explain the absence of a central nervous system or skeletal muscle phenotype in Pax7-/- mice at birth.5,6,7
While Pax3 plays a critical role during development, Pax7 becomes the predominant factor during postnatal muscle growth and regeneration. In postnatal muscles, myogenic progenitor cells begin to localize to their niche between the basal lamina and the muscle fiber.8 At this stage, the cells are named satellite cells, based on this specific anatomical location. At birth, satellite cells represent approximately 30% of sublaminar muscle nuclei in mice; this decreases to <5% in two-month-old adult mice.9 This decline reflects fusion of satellite cells to muscle fibers during postnatal muscle growth. In resting adult muscle, satellite cells are mitotically quiescent, but they are poised for activation. After an injury, satellite cells re-enter cell cycle and become myoblasts, which undergo multiple rounds of cell division before differentiating and fusing into myofibers.10 Expression of Pax7 is confined to quiescent satellite cells and proliferating myoblasts and is downregulated during terminal differentiation.11 Deletion of Pax7 in primary myoblasts in vitro results in cell cycle arrest and precocious differentiation. Accordingly, mice carrying a targeted null variant of Pax7 (Pax7-/-) or conditionally inducible alleles showed that deletion of Pax7 impairs satellite cell survival leading to the loss of the satellite cell pool and ablated skeletal muscle regeneration.11,12,13 Pax3 expression is not able to compensate the antiapoptotic role of Pax7 in order to maintain the population of satellite cells in postnatal muscles, even though Pax3 remains expressed in a population of satellite cells in some postnatal muscles.14 Overall, animal studies revealed that the role of Pax7 during skeletal muscle development is limited (masked by Pax3), but it is irreplaceable postnatally for satellite cell survival and skeletal muscle regeneration.
Here, we identified, for the first time, five individuals from four unrelated families with homozygous variants predicted to cause loss of function in PAX7, leading to a myopathy of variable severity. Contrary to other myopathies, such as muscular dystrophies, which induce myofiber fragility and muscle degeneration, our findings indicate that PAX7 variants do not affect myofiber stability, but rather lead to the exhaustion of the satellite cell pool and consequently decreased muscle growth and regeneration capacity.
MATERIALS AND METHODS
Patients
Patients were evaluated in different hospitals in Canada, Germany, Palestine, and Saudi Arabia. All individuals or their guardians gave written informed consent before undergoing evaluation, in agreement with the Declaration of Helsinki. In addition to clinical evaluation, tissue samples were collected, when appropriate, for diagnostic purposes; the remaining samples were used for research purposes. The participants and/or the parents gave written informed consent to participate in the research study, which was approved by the ethics boards of the respective institutions (individual 1: CHUSJ 2016–962; individual 2: TUM 2341/09; individual 3: local IRB vote 180/2010BO1; individuals 4–5: Psmmc 943/2017). Written informed consent was provided for images of patients appearing in this paper.
Genetics
We performed solo exome sequencing to individuals’ DNA extracted from peripheral blood.15 In brief, coding regions were enriched using a SureSelect Human All Exon V5 kit (Agilent) or TruSeq (Illumina) followed by sequencing as 100-bp paired-end runs on an Illumina HiSeq. Reads were aligned to the human reference genome (UCSC Genome Browser build hg19) using Burrows–Wheeler Aligner (v.0.7.5a).16 Single-nucleotide variants and small insertions and deletions (indels) were detected with SAMtools (v.0.1.19). Variants were first checked for genes known to be associated with neurological disorders. Next variants were filtered for homozygous or compound heterozygous changes and prioritized according to frequency in public databases, conservation, and prediction scores. Sanger sequencing was used to confirm the identified variants and test the carrier status of unaffected family members.
Immunohistochemistry
Immunohistochemistry was performed as previously described.17 The following antibodies were used: PAX7 (DSHB), Desmin (D33, Agilent, Dako), MYF5 (C-20, Santa Cruz), MYH3 (F1.652, DSHB), MyHC fast (a474, DSHB), Dystrophin (ab15277, Abcam), and H3P (06–570, Upstate). Pictures were taken with a Leica inverted microscope and analyzed using ImageJ software. Evaluators were blinded to the identity of the sample.
RNA isolation, retrotranscription, and quantitative real- time PCR
Total RNA was extracted from patients’ samples using commercial kits (Qiagen). Retrotranscription and real-time polymerase chain reaction (PCR) were performed using the CFX96 touch real-time PCR detection system (Biorad), and gene expression analysis was performed with the CFX manager software. Primers are indicated in Suppl. Table S1. All data were corrected against a combination of housekeeping genes (18S and GAPDH) as internal control. Experiments were replicated at least twice in the laboratory in technical triplicates.
RESULTS
Genetics
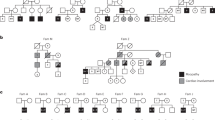
We present five individuals with myopathy from four unrelated families of consanguineous parents (Fig. 1a). Patients 1, 2, and 3 were collated using GeneMatcher,18 while patients 4 and 5 were recruited by screening the Centogene database after the link between PAX7 and myopathy was established. Exome sequencing and bioinformatics data analyses have been performed in four different centers. Rare variants in known genes associated with myopathy were not identified in any of the individuals. Under the assumption of autosomal recessive inheritance and based on the predicted detrimental functional impact of the variants as well as known physiological function of the gene product, the PAX7 (NM_002584.2) changes were independently prioritized as putatively disease-causing candidate variants (Table 1). We identified four different PAX7 disease alleles (Fig. 1b) including two homozygous stop variants (individual 1: c.433 C>T, p.[Arg145*]; individual 3: c.220 C>T, p.[Arg74*]), a homozygous missense variant (individuals 4 and 5: c.166 C>T, p.[Arg56Cys]), and a homozygous splice site variant (individual 2: c.86–1 G>A). All four variants are absent from or extremely rare in public databases (all absent from ExAC, minor allele frequency [MAF] <0.001% in gnomAD; Table 1) and affect highly conserved residues (Fig. 1c, and Suppl. Fig. S2). Segregation, prediction, and conservation scores are in concordance with pathological features of altered PAX7. CADD scores of the variants are between 25 and 46. Further, the missense variant c.166 C>T, p.(Arg56Cys) is consistently predicted to be pathogenic by MutationTaster, Provean, PolyPhen-2, and SIFT (Table 1) and is highly conserved (PhyloP_100way 6.0, Fig. 1c, Suppl. Fig. S2). PAX7 is well conserved in the general population and seems to be intolerant to loss-of-function variants. According to the ExAC database, 15.9 loss-of-function variants are expected, but only 3 variants are found (probability of loss-of-function intolerance [pLI] score of 0.63), all located in exon 8, and none in homozygosity.

Identification of homozygous PAX7 variants in five affected individuals from four consanguineous families. (a) Pedigrees of families from Canada (individual 1), Germany (individual 2), Palestine (individual 3), and Saudi Arabia (individuals 4 and 5). The affected individuals carry homozygous variants in PAX7. Open symbols represent unaffected individuals, and filled symbols represent affected individuals. Black arrows indicate the affected individuals discussed in the text. (b) Position of the four identified homozygous variants in the PAX7 gene. PAX7 messenger RNA (mRNA) is spliced into three different isoforms that differ by their N-terminal protein sequence and the presence of an OAR domain in isoform 3. Gray, untranslated region (UTR); broad bars, translated sequence; thin gray lines, intron (not drawn to scale); turquoise, paired box domain; green, homeodomain; dark blue, exon 8 sequence specific for isoforms 1 and 2; pink, termination codon; orange, OAR domain; red, variants identified in this study. (c) Alignment of amino acids or nucleic acids flanking the different variants in the PAX7 gene show high evolutionary conservation. The variants are located upstream or within the paired box domain, which is highly conserved across species. Gray box indicates variation in amino acid residues.
Clinical features
Every individual in our cohort had a normal prenatal history, except for individual 2, who was noted to have diminished fetal movement (Table 2). Perinatal history was unremarkable in individuals 3 to 5, but individuals 1 and 2 showed clinical signs of myopathy at birth. Individual 1 was born at 41 weeks of gestation and was noted to have inefficient suck-swallowing mechanisms requiring tube feeding from birth. Individual 2 was born at 39 weeks of gestation and showed signs of myasthenia after sepsis and cyanosis with increased plasma lactate, which is a common feature of diseases affecting the mitochondrial energy metabolism. However, no acetylcholine antibodies were detected, metabolic screening was negative, and no signs of impaired respiratory chain enzyme activities were observed.
The first clinical signs were observed in the first months of life, which included axial or generalized hypotonia. Skeletal muscle atrophy was observed in every individual, affecting predominantly the trunk/neck muscles and/or lower limbs and distal extremities. Postnatal growth was symmetric, but most individuals presented with short stature or growth deficit (Table 2). All patients developed scoliosis ranging from moderate to severe. Gait was affected in individual 1 (wheelchair bound), individual 3 (preserved with difficulties), individual 4 (waddling gait), and individual 5 (walking with difficulties on tiptoe and heel walking), but was considered normal in individual 2. The oldest individuals in our cohort were experiencing disease progression. For instance, individual 1 was able to walk with a walker until 6 years old; he then became wheelchair bound and is now bedridden (at 14 years old) (Fig. 2). In addition, symptoms of progressive respiratory insufficiency due to muscle weakness developed, which led to treatment with bilevel positive airway pressure (BiPAP) at 4.5 years, initially at night only, later continuously. Progression in disease severity was also observed in individual 3 who died prematurely at the age of 7. Although the cause of death is unknown, the family history revealed three similar affected siblings with reported muscle hypotonia and weakness, who all died within the first decade of life, suggesting the disease as the likely cause of death. Progression in the severity of the scoliosis was also observed in patients 4 and 5, going from a Cobb angle of 40° at 2.7 years old to 60° at 4 years old.

Physical characteristics of individuals affected by PAX7 variants. Photographs of the affected individuals at different time points showing facial dysmorphic features, muscle atrophy, joint contracture, and scoliosis.
Ptosis is a common feature and was found in all probands except individual 3. Dysmorphic facial features are variable and range from isolated triangular facies (individuals 4 and 5) to more severe (dolichocephaly, plagiocephaly, micrognathia with a high-arched palate, posteriorly rotated ears, and prominent nasal root in individual 1; and low-set ears, hypotonic facies, high palate, flat nose, and facial hirsutism in individual 2) (Fig. 2 and Table 2). Cognitive development, socialization, and behavior are normal in all patients.
Laboratory tests and exams
For individual 1, laboratory tests including creatine kinase (CK), ammonia, plasma amino acids, urine organic acids, transferrin isoelectric focusing, fragile X, karyotype, comparative genomic hybridization (CGH) array, and DMPK analysis for myotonic dystrophy type 1 (MIM 160900) were normal. Normal CK levels were also noted in every other individual tested, suggesting the absence of muscle degeneration/injury. Electromyograph (EMG) exam for individual 1 was normal at 3 months old and 2 years old, but showed a nonspecific pattern suggestive of a myopathy at 13.5 years old (Table 2). EMG exam was normal in every other patient tested, suggesting that the disease is not of neuromuscular or neuropathic origin. Echocardiography and brain magnetic resonance imaging (MRI) studies were normal in every patient tested.
Skeletal muscle biopsy analysis
Muscle biopsies were obtained for diagnostic purposes in individuals 1, 2, and 4 and reviewed by neuropathologists. For individual 1, a first biopsy performed on the gracilis muscle at 3 months of age showed mild variability in size and shape of muscle fibers without diagnostic neurogenic or myopathic findings on hematoxylin–eosin (H&E) (Fig. 3a, left panel) and on a full muscle biopsy panel including histochemical stains, histoenzymology, and electron microscopy (not shown). Given the progression of the disease, a second gracilis muscle biopsy was performed at 3.7 years of age and showed more definite pathology including wide patches of fatty replacement, focal endomysial fibrosis, and scattered angulated atrophic fibers (Fig. 3a, middle panel). Fiber typing by immunohistochemistry showed that the angulated atrophic fibers were of both type I and type II, without fiber type grouping—a pattern suggestive but not diagnostic of neurogenic atrophy (Suppl. Fig. S3A). Of note, the fatty replacement was in excess compared with the degree of fibrosis and no conspicuous dystrophic changes such as fiber rounding or necrotic/regenerative fibers were seen. The full muscle biopsy panel, including NADH-TR and Gomori trichrome and electron microscopy did not reveal additional diagnostic features (Suppl. Fig. S3B,C). A sural nerve biopsy performed simultaneously was within normal limits for the age (Suppl. Fig. S4). Muscle biopsy of individual 2 performed on quadriceps at 2.3 years of age showed no significant abnormalities on H&E (Fig. 3a, right panel) and other routine stains including Gomori trichrome, COX/SDH, and NADH-TR (Suppl. Fig S5). Biochemical assay of OXPHOS enzymes activity showed no diagnostic mitochondrial dysfunction (Suppl. Fig S6). A muscle biopsy of the vastus lateralis muscle of individual 4 was performed at 3 years of age (not shown). Atrophic fibers were observed, predominantly for type IIB fibers. The connective tissue elements were unremarkable. A few regenerating myofibers were observed, but no necrotic myofibers were detected. ATPase staining revealed type I fiber preponderance and small zone of fiber grouping, suggesting slight denervation and reinnervation.

Analysis of muscle biopsies from patients carrying PAX7 variants. (a) Photomicrograph of gracilis muscle biopsy performed at 3 months of age (left panel; hematoxylin and eosin [H&E]) and 3.7 years of age (middle panel; H&E, saffron) on individual 1. Asterisk indicates marked fatty replacement, black arrows show focal endomysial fibrosis without fiber rounding or necrosis, and open arrows point to scattered atrophic angulated fibers suggesting possible denervation. Right panel shows a picture of H&E stain of quadriceps muscle biopsy performed at 2.3 years of age on individual 2. Scale bars (50 µm for left panel and 100 µm for middle and right panels). (b) Real-time quantitative polymerase chain reaction (qPCR) quantification of RNA isolated from muscle biopsies for the myogenic factors PAX7, PAX3, MYF5, MYF6, MYOD1, MYOG, MYH3, and DES. Graphs indicate the fold change (mean +/- SEM) in expression for individual 1 compared with 4 age-matched healthy controls, and with 10 age-matched healthy controls for individual 2. (c–h) Immunofluorescence or immunohistochemical staining of muscle section from individuals with PAX7 variants (right column) and healthy age-matched individual (left column). (c) Immunofluorescence staining for PAX7 (red); wheat germ agglutinin (WGA, green) a marker of connective tissue; and DAPI (nuclei in blue). White arrowheads indicate PAX7-expressing satellite cells. Dashed boxes are zoomed in and displayed in the right corner of the picture. (d) Immunohistochemical staining for MYF5 (orange-brown cells) with hematoxylin counterstain (nuclei in blue). Black arrowheads indicate MYF5-expressing myogenic cells. (e) Immunofluorescence staining for desmin (green) and DAPI (blue). White arrowheads indicate desmin-positive myoblasts. (f) Immunohistochemical staining for H3P (orange-brown cells) with hematoxylin counterstain (nuclei in blue). While most nuclei are positive in the controls, only a few positive nuclei are visible in the PAX7-mutant patient, which are identified by black arrowheads. (g) Immunofluorescence staining for MYH3 (red), WGA (green), and DAPI (blue). White arrowheads indicate MYH3-positive regenerative myofibers. (h) Immunofluorescence staining for dystrophin (DMD, red) and DAPI (nuclei in blue). (c’–h’) Quantification of the different myogenic stains. n = 2–4 healthy controls. Graphics are presented as mean +/- SEM. ND not detected.
Expression analysis of myogenic regulatory factors
Quantitative PCR was performed on muscle tissue of individuals 1 and 2. Analyses revealed a strongly decreased amount of PAX7 transcript in individual 1 (not detectable) and individual 2 (15-fold decrease) compared with age-matched controls (Fig. 3b). A similar decrease in the expression levels of MYF5, a downstream target of PAX7,19 was observed in individual 1 (not detectable) and individual 2 (22-fold decrease). In contrast, the expression level of PAX3 was increased in individuals 1 and 2 (twofold and tenfold, respectively). The expression level of other myogenic factors (MYF6, MYOD1, MYOG, DES) differed between the individuals; individual 1 displayed a moderate decrease in the expression of these factors, while they were moderately increased in individual 2. Finally, the expression of embryonic myosin heavy chain (MYH3) was elevated in both individuals, suggesting active muscle fiber regeneration.
Immunostaining for myogenic cells
Immunostaining confirmed a complete absence of PAX7-expressing and MYF5-expressing cells in individuals 1 and 2 (Fig. 3c, d’ and Suppl. Fig. S7). The presence of activated myogenic cells expressing desmin, a myoblast marker, was observed although their number is strongly reduced compared with controls (Fig. 3e–e’). A marked reduction in the number of cells positive for phosphohistone H3 (H3P), a mitosis marker, was found in absence of PAX7 (Fig. 3f–f’). Myofibers expressing MYH3 were observed only in the muscle of the patients (not in healthy controls) indicating the presence of ongoing muscle regeneration (Fig. 3g–g’). The number of nuclei per myofiber was reduced in the absence of PAX7, suggesting a lack of myogenic cells that fuse to donate their nuclei to the myofibers (Fig. 3h–h’).
DISCUSSION
In this paper, we provide evidence that biallelic variants predicted to cause loss of function in PAX7, the master regulator of satellite cell survival, are a novel genetic cause of myopathy in humans. Multiple classes of myopathies have been described including muscular dystrophies, mitochondrial myopathies, metabolic myopathies, and others. Contrary to these types of myopathies, our findings indicate that predicted loss-of-function variants in PAX7 do not directly affect the myofiber structure and/or function, but rather lead to the exhaustion of the satellite cell pool. Analysis of muscle biopsies revealed no signs of myofiber degeneration; however, the lack of satellite cells leads to impaired muscle growth and fibroadipose tissue replacement, which results in progressive muscle weakness. To our knowledge, this study is the first to identify a genetic cause of myopathy that specifically affects muscle stem cells.
A previous case study identified individuals from a consanguineous family with PAXBP1 variant, which encodes for an adapter protein linking PAX3 and PAX7 to the histone methylation machinery.20 The phenotype of the patients was characterized by global developmental delay, severe intellectual disabilities, and severe hypotonia. Because we did not observe cognitive disabilities in our patient cohort, it is likely that this phenotype is related to the role of PAXBP1 in PAX3 functioning. Furthermore, one prior case study was published describing the clinical presentation of two brothers with a homozygous PAX7 variant in intron 8 (ref. 21). The phenotype of the two brothers shows some similarities to our patient cohort, such as axial hypotonia and postnatal growth retardation; however, they also exhibit severe cognitive and behavioral disabilities that were not observed in our individuals. This phenotypic difference could be explained by many factors. First, in the previous study the variant in intron 8 is localized 5’ of exon 9, which is only included in isoform 3 of PAX7, but not in isoforms 1 and 2. No analysis of PAX7 expression was performed on the patient’s muscle; however, in vitro studies suggested abnormal splicing of isoform 3 with loss of the OAR domain and accumulation of PAX7 isoform 3 in the cell nucleus. Furthermore, both children also had a homozygous missense variant in the KIF21A motor domain, which was predicted to be pathogenic. In contrast to PAX7, KIF21A is highly expressed in the central nervous system (GTEx Portal November 2017). Depletion of KIF21A causes aberrant microtubule formation in vitro and homozygous knockout mice die within 24 hours of life.22,23 The possible effect of this homozygous missense variant was not further elucidated. However, two possible mechanisms come to mind to explain the phenotypic differences between our group of patients and the two brothers described previously: either the divergent phenotype in the brothers is caused by accumulation of aberrant PAX7 protein compared with absent PAX7 protein in our cohort, and/or the brothers have in fact two autosomal recessive conditions, the second caused by a KIF21A variant with a new phenotype causing the severe developmental disability and behavioral difficulties not observed in our individuals. Contrary to this previous publication, the present paper allows to clearly characterize this myopathy and establish causality because (1) the findings are from multiple unrelated individuals, (2) the pathogenic variants are located within or upstream the highly conserved paired box domain, and (3) muscle samples analysis confirmed the lack of PAX7, which is associated with satellite cell exhaustion and impaired muscle growth.
Expression analysis also revealed a severe reduction in MYF5 expression, which is consistent with the ability of PAX7 to target MYF5.19 A recent study showed that individuals with biallelic homozygous loss-of-function variants in MYF5 have clinical features characterized by external ophthalmoplegia, ptosis, and rib and vertebral anomalies (including scoliosis); but, contrary to our patient cohort, they show no sign of hypotonia or muscle weakness.24 This discrepancy is likely caused by the functional redundancy of MYF5 with another myogenic regulatory factor, MYOD1, which was demonstrated in various mouse models to be able to largely compensate for the lack of MYF5.25,26
The role of Pax7 in skeletal muscle has been widely studied in animal models. Studies using Pax7-null mice demonstrated that this factor is dispensable during embryonic development, but is absolutely required in postnatal muscles.11 Conditional ablation mouse models confirmed that Pax7 is indispensable for satellite cell function and muscle regeneration in the adult.13,27 In addition to its role in muscle regeneration, it was shown using a conditional depletion mouse model that Pax7-expressing satellite cells participate in muscle growth and hypertrophy in growing mice but not in fully grown mice.27,28,29 Findings in Pax7-deficient mice correlate with the muscle phenotype observed in our patient cohort, which displayed normal prenatal muscle development, but impaired postnatal muscle growth and progressive muscle weakness.
Our clinical findings indicate that the five individuals have similar symptoms although the severity of the disease varies. Quantitative PCR (qPCR) analysis suggests that the degree of downregulation of PAX7 expression could partially explain this interindividual variability. The most severely affected patient (individual 1) has a complete absence of PAX7 expression, which suggests nonsense-mediated decay, while individual 2, who has a milder phenotype, has a strongly reduced (15-fold decrease) but not complete lack of PAX7 expression. However, immunohistochemical findings showed that PAX7 could not be detected at the protein level in either of these individuals. Unfortunately, we were not able to assess PAX7 expression in patients 4 or 5; however, we have strong evidence that supports the pathogenic relevance of the PAX7 missense variant p.(Arg56Cys). First, it has high pathogenicity prediction scores and it is absent from public databases (Table 1). Moreover, this arginine residue has a high phylogenetic conservation within orthologous PAX7 proteins (Fig. 1c, Suppl. Fig S2); and it is highly conserved within all PAX family members in the human genome (Suppl. Fig. S8A). Analysis of the crystalline structure of the human PAX6 protein30 shows that the corresponding arginine residue (p.Arg26 in PAX6) is part of the helix-turn-helix (HTH) domain superfamily, which interacts with DNA (Suppl. Fig. S8B–E). A detailed study on the corresponding variant in PAX6 showed that the mutated protein failed to bind DNA through its paired domain.31 Because the HTH domain is highly conserved within PAX genes, further evidence for pathogenic relevance of the PAX7 missense variant comes from diseases caused by the corresponding variants in other PAX genes, such as PAX3 (p.Arg56Leu),32 PAX6 (p.Arg26Gly),33 PAX8 (p.Arg31His),34 and PAX9 (p.Arg26Trp).35
Similar to our patient cohort, variable phenotypes were described in Pax7-/- mice from different mouse strains suggesting an important influence of genetic modifiers on phenotype severity. Pax7-/- mice in a C57/BL6 background appear normal at birth but fail to thrive and die within 3 weeks postnatal,11 while Pax7-/- mice in a 129 Sv/J background are able to live up to 6 months, although they have reduced body weight and muscle mass.12 The persistence of a population of myogenic progenitor cells might partially contribute to mitigate the myopathy caused by the absence of Pax7. For instance, a low proportion of Pax7-/- mice on a mixed C57/BL6/129 Sv background are able to survive until adulthood. In these surviving mice, a small population of myogenic progenitor cells was observed at P11 but was completely exhausted at P60.36 Notably, in Pax7-/- mice in a 129 Sv/J background, which have higher survival potential, the presence of interstitial Pax3-expressing cells was noted to persist until adulthood (up to 6 months).12 Similarly, the muscle biopsies of individuals of our patient cohort revealed the presence of a small population of desmin-expressing myoblasts and MYH3-expressing myofibers, which indicate the persistence of a certain myogenic potential in human in the absence of PAX7. Although the antiapoptotic role of Pax3 is limited in postnatal muscle,14 it can be hypothesized that the overexpression of PAX3 levels observed in the muscles of our patients could contribute to preserve a subset of myogenic cells.37
Other cell types with myogenic potential have also been identified, such as side-population cells, pericytes, and PDGFRα+-mesenchymal progenitors.8 The myogenic capacity of these cell types has been mostly studied in vitro and requires the presence of satellite cells to mediate their myogenic potential. Other cell types, such as twist2-positive cells and smooth muscle mesenchymal cells, were also recently identified as cells that could contribute to myogenesis in a Pax7-independent manner.38,39 The hypothesis that alternate sources of myogenic cells contribute to mitigate the disease is supported by our qPCR data showing that individual 2, who has a milder phenotype, expresses higher levels of myogenic regulatory factors (PAX3, MYF6, MYOD1, MYOG) compared with individual 1, who has a more severe phenotype. Considering that the disease is progressive, these differences could be explained by the age at which the biopsies were performed (severe phenotype for individual 1 at 3.7 years and mild phenotype for individual 2 at 2.3 years). Nevertheless, despite the potential contribution of nonsatellite cells to myogenesis, animal studies using conventional or conditional Pax7 knockout clearly demonstrated that satellite cells are absolutely required for muscle growth and regeneration.13,27,40
In conclusion, our findings from multiple unrelated individuals are the first to clearly establish causality between pathogenic variants in PAX7 and a new genetic cause of myopathy. Prenatal muscle development appears normal in our patient cohort; however, the variants provoke the progressive exhaustion of the satellite cell pool postnatally, which leads to myofiber atrophy and fibroadipose tissue replacement. The ability of the patients to maintain a population of myogenic cells could partially contribute to mitigating the disease; however, the disease progression suggests that this myogenic population is depleted over time and/or is unable to compensate for the absence of satellite cells to ensure long-term muscle growth. Overall, contrary to other types of myopathies, such as muscular dystrophies, our findings have identified a completely new class of myopathy that does not directly affect muscle fibers but rather impairs satellite cell survival, leading to reduced skeletal muscle growth and regeneration.
References
Chi N, Epstein JA. Getting your Pax straight: Pax proteins in development and disease. Trends Genet. 2002;18:41–47.
Blake JA, Ziman MR. Pax genes: regulators of lineage specification and progenitor cell maintenance. Development. 2014;141:737–751.
Marshall AD, Grosveld GC. Alveolar rhabdomyosarcoma—the molecular drivers of PAX3/7-FOXO1-induced tumorigenesis. Skelet Muscle. 2012;2:25.
Mansouri A, Stoykova A, Torres M, Gruss P. Dysgenesis of cephalic neural crest derivatives in Pax7-/- mutant mice. Development. 1996;122:831–838.
Goulding MD, Chalepakis G, Deutsch U, Erselius JR, Gruss P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991;10:1135–1147.
Jostes B, Walther C, Gruss P. The murine paired box gene, Pax7, is expressed specifically during the development of the nervous and muscular system. Mech Dev. 1990;33:27–37.
Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435:948–953.
Dumont NA, Bentzinger CF, Sincennes M-C, Rudnicki MA. Satellite cells and skeletal muscle regeneration. Compr Physiol. 2015;5:1027–1059.
Bischoff R. The satellite cell and muscle regeneration. In: Engel AG, Franzini-Armstrong C, eds. Myology. 2nd edn. New York: McGraw-Hill; 1994. p. 97–118.
Dumont NA, Wang YX, Rudnicki MA. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development. 2015;142:1572–1581.
Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786.
Kuang S, Chargé SB, Seale P, Huh M, Rudnicki MA. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol. 2006;172:103–113.
von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci U S A. 2013;110:16474–16479.
Relaix F, Montarras D, Zaffran S, et al. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J Cell Biol. 2006;172:91–102.
Kremer LS, Danhauser K, Herebian D, et al. NAXE mutations disrupt the cellular NAD(P)HX repair system and cause a lethal neurometabolic disorder of early childhood. Am J Hum Genet. 2016;99:894–902.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760.
Dumont N, Rudnicki M. Characterizing satellite cells and myogenic progenitors during skeletal muscle regeneration. In: Pellicciari C, Biggiogera M, eds. Histochemistry of single molecules. Methods in molecular biology. New York: Springer; 2017. p. 179–188.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–930.
McKinnell IW, Ishibashi J, Le Grand F, et al. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat Cell Biol. 2008;10:77–84.
Alharby E, Albalawi AM, Nasir A, et al. A homozygous potentially pathogenic variant in the PAXBP1 gene in a large family with global developmental delay and myopathic hypotonia. Clin Genet. 2017;92:579–586.
Proskorovski-Ohayon R, Kadir R, Michalowski A, et al. PAX7 mutation in a syndrome of failure to thrive, hypotonia, and global neurodevelopmental delay. Hum Mutat. 2017;38:1671–1683.
van der Vaart B, van Riel WE, Doodhi H, et al. CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev Cell. 2013;27:145–160.
Cheng L, Desai J, Miranda CJ, et al. Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron. 2014;82:334–349.
Di Gioia SA, Shaaban S, Tüysüz B. Recessive MYF5 mutations cause external ophthalmoplegia, rib, and vertebral anomalies. Am J Hum Genet. 2018;103:115–124.
Yamamoto M, Legendre NP, Biswas AA, et al. Loss of MyoD and Myf5 in skeletal muscle stem cells results in altered myogenic programming and failed regeneration. Stem Cell Rep. 2018;10:956–969.
Rudnicki MA, Schnegelsberg PNJ, Stead RH, Braun T, Arnold H-H, Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359.
Sambasivan R, Yao R, Kissenpfennig A, et al. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–3656.
Fry CS, Lee JD, Mula J, et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med. 2015;21:76–80.
Murach KA, White SH, Wen Y, et al. Differential requirement for satellite cells during overload-induced muscle hypertrophy in growing versus mature mice. Skelet Muscle. 2017;7:14.
Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO. Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev. 1999;13:1263–1275.
Singh S, Stellrecht CM, Tang HK, Saunders GF. Modulation of PAX6 homeodomain function by the paired domain. J Biol Chem. 2000;275:17306–17313.
Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin CT. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I). Am J Hum Genet. 1993;52:455–462.
Hanson IM, Fletcher JM, Jordan T, et al. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat Genet. 1994;6:168–173.
Macchia PE, Lapi P, Krude H, et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet. 1998;19:83–86.
Lammi L, Halonen K, Pirinen S, Thesleff I, Arte S, Nieminen P. A missense mutation in PAX9 in a family with distinct phenotype of oligodontia. Eur J Hum Genet. 2003;11:866–871.
Oustanina S, Hause G, Braun T. Pax7 directs postnatal renewal and propagation of myogenic satellite cells but not their specification. EMBO J. 2004;23:3430–3439.
Hirai H, Verma M, Watanabe S, Tastad C, Asakura Y, Asakura A. MyoD regulates apoptosis of myoblasts through microRNA-mediated down-regulation of Pax3. J Cell Biol. 2010;191:347–365.
Liu N, Garry GA, Li S, et al. A Twist2-dependent progenitor cell contributes to adult skeletal muscle. Nat Cell Biol. 2017;19:202–213.
Giordani L, He GJ, Negroni E, et al. High-Dimensional Single-Cell Cartography Reveals Novel Skeletal Muscle-Resident Cell Populations. Mol Cell. 2019;74:609–621.
Lepper C, Partridge TA, Fan C-M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138:3639–3646.
Acknowledgements
We thank the participants and their families for their participation in this study. This study was supported by the German Bundesministerium für Bildung und Forschung through the Juniorverbund in der Systemmedizin “mitOmics” (FKZ01ZX1405C to T.B.H.) and Horizon2020 through the E-Rare project GENOMIT (01GM1603 and 01GM1207 for H.P. and FWFI2741B26 for J.A.M.) and the Deutsche Forschungsgemeinschaft (SCHO754/52 to L.S. and BA2427/22 to P.B.) as well as the Vereinigung zur Förderung Pädiatrischer Forschung und Fortbildung Salzburg, the EU FP7 Mitochondrial European Educational Training Project (317433 to H.P. and J.A.M.), and the EU Horizon2020 Collaborative Research Project SOUND (633974 to H.P.). N.A.D. is supported by grants from the Fonds de recherche du Québec–Santé (35015), Canadian Institutes of Health Research (388296), Rare Disease Foundation (2301), and CHU Sainte-Justine Foundation. N.A.D. acknowledges the support of ThéCell and Stem Cell Network.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Feichtinger, R.G., Mucha, B.E., Hengel, H. et al. Biallelic variants in the transcription factor PAX7 are a new genetic cause of myopathy. Genet Med 21, 2521–2531 (2019). https://doi.org/10.1038/s41436-019-0532-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0532-z
Keywords
This article is cited by
-
The identification of PAX7 variants and a potential role of muscle development dysfunction in congenital scoliosis
Cell Regeneration (2022)
-
Muscle stem cell adaptations to cellular and environmental stress
Skeletal Muscle (2022)
-
Pax7 as molecular switch regulating early and advanced stages of myogenic mouse ESC differentiation in teratomas
Stem Cell Research & Therapy (2020)
-
First-line exome sequencing in Palestinian and Israeli Arabs with neurological disorders is efficient and facilitates disease gene discovery
European Journal of Human Genetics (2020)
-
Human muscle-derived CLEC14A-positive cells regenerate muscle independent of PAX7
Nature Communications (2019)
