
Abstract
Purpose
Prenatal genetic carrier screening can identify parents at risk of having a child affected by a recessive condition. However, the conditions/genes most appropriate for screening remain a matter of debate. Estimates of carrier rates across genes are needed to guide construction of carrier screening panels.
Method
We leveraged an exome sequencing database (n = 123,136) to estimate carrier rates across six major ancestries for 415 genes associated with severe recessive conditions.
Results
We found that 32.6% (East Asian) to 62.9% (Ashkenazi Jewish) of individuals are variant carriers in at least one of the 415 genes. For couples, screening all 415 genes would identify 0.17–2.52% of couples as being at risk for having a child affected by one of these conditions. Screening just the 40 genes with carrier rate >1.0% would identify more than 76% of these at-risk couples. An ancestry-specific panel designed to capture genes with carrier rates >1.0% would include 5 to 28 genes, while a comparable panethnic panel would include 40 genes.
Conclusion
Our work guides the design of carrier screening panels and provides data to assist in counseling prospective parents. Our results highlight a high cumulative carrier rate across genes, underscoring the need for careful selection of genes for screening.
Similar content being viewed by others

INTRODUCTION
Prenatal genetic carrier screening (PGCS) has changed rapidly over the past few decades, driven by advances in technology, increased awareness of rare inherited conditions and their impact on families, and increased availability of treatments for inherited rare diseases. The model has moved from screening only a limited number of variants (e.g., ∆508 in CFTR) in a small handful of conditions (e.g., CFTR for cystic fibrosis) in only high-risk populations (e.g., individuals of Caucasian ancestry) to screening many variants in a large number of genes across broad ancestry groups.1,2 The latter screening paradigm has been called expanded carrier screening (ECS). ECS leverages next-generation sequencing or high-throughput genotyping to allow simultaneous and affordable assessment of genetic variation of a panel of genes. However, it is unclear which genes and variants should be included on ECS panels. Similarly, it is unclear whether individuals of all ancestries should receive the same screening panel or whether separate panels of genes should be tested for each ancestry.
The costs and potential benefits should be considered when deciding which genes should be included as part of an ECS panel. The cost of adding genes to a panel comes in two quantifiable forms. The first is the technical cost, which is becoming increasingly negligible in the era of next-generation sequencing.3 The second quantifiable cost comes from downstream interpretation and counseling. Adding genes to a panel will increase the number of variant carriers identified, which will necessitate additional counseling as well as confirmatory and follow-up testing, including testing of the patient’s partner. A third cost, which is very challenging to quantify, is the cost of anxiety to the patient. Identifying carriers of pathogenic variants from an ECS panel potentially increases stress and anxiety for these individuals, though existing evidence suggests that there may not be much anxiety provoked by genetic testing.4,5 The benefit of adding genes to a panel is that it can increase identification of at-risk couples, which can inform preconception decisions and decisions regarding management of an established pregnancy.
Professional organizations including the American College of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics and Genomics (ACMG) have made recommendations regarding the genes and conditions for which prenatal screening can or should be performed. ACOG recommended a lower limit carrier frequency (1/100) for screening conditions; however, this threshold is not based on empirical evidence.6 These recommendations have implications for financial stakeholders and public health given the approximately 4 million births in the United States annually. Therefore, additional data is needed to inform professional organization screening guidelines.
Cost and benefit of ECS cannot be determined until conditions appropriate for screening are better understood from a population perspective. In this study, we leveraged a population database with existing sequencing data to estimate the carrier rates for all severe recessive Mendelian conditions to inform the design and utility of ECS panels. We also sought to use this data to understand how the number of conditions screened influences the proportion of couples impacted.
MATERIALS AND METHODS
Study population
We used data from gnomAD v2.0.2, which is comprised of summary-level data for 123,136 exome sequencing samples.7 The data was downloaded from http://gnomad.broadinstitute.org. For each variant in gnomAD, the allele count, allele number, and number of individuals who are homozygotes are provided for each of the ancestry groups. No individual-level data are available.
Variants and genes analyzed
To obtain a list of variants to analyze, we downloaded the ClinVar database.8,9 We used a version of the ClinVar database that was parsed to facilitate mapping of variants to genes and conditions.10 We included all variants that are annotated as either likely pathogenic or pathogenic; variants with conflicting annotations were not included.11 We did not filter variants with regard to their rating status. A set of 19 variants (Table S1) were excluded from the analyses because they are common (minor allele frequency [MAF] > 0.5% in at least one ancestry) and are of known low penetrance, were excluded in a prior paper,12 or have poor sequencing quality in gnomAD.
We extracted a list of 924 genes previously annotated as being associated with severe Mendelian conditions.13 Among these 924 genes, we included the 416 genes that are annotated as acting in an autosomal recessive manner.8 We also excluded ABCC6 since the gene had poor sequencing quality in gnomAD. This resulted in a final list of 415 genes analyzed. A total of 15,797 variants across these 415 genes were analyzed.
Estimation of gene carrier rate (GCR)
We first calculated the variant carrier rate (VCR) for each pathogenic or likely pathogenic variant:
Here AC is the total allele count for the variant, Hom is the number of individuals who are homozygous for the variant, and AN is the total number of alleles analyzed for the variant.
The GCR for a gene g can then be estimated as:
Here VCRi is the variant carrier rate for variant i, and v is the number of variants of interest in gene g.
These calculations were performed separately for each ancestry.
Estimation of cumulative carrier rate (CCR)
The CCR for a set of genes s can be estimated as:
Here GCRi is the gene carrier rate for gene i in a set of s genes. These calculations were performed separately for each ancestry.
Estimation of at-risk couple rates
We also calculated the at-risk couple rate (ACR), which is the estimated proportion of couples who each carry a likely pathogenic or pathogenic variant in the same gene. In contrast to prior work that calculated the proportion of affected offspring,12 we eschewed this metric as our primary metric because the proportion of affected offspring is dependent on penetrance. The at-risk couple rate ACR can be estimated as:
Here GCRi,a1 is the GCR for gene i in ancestry 1 and GCRi,a2 is the GCR for gene i in ancestry 2. This calculation was performed for all s genes in a set of genes and across all unique pairwise combinations of a1 and a2.
Software
Processing of ClinVar data was performed using custom Python scripts (v2.7) and Bcftools v1.2 (ref. 14). Analyses and plotting were performed using R v3.1. All code for this paper is available at https://github.com/mhguo1/ECS/.
RESULTS
Study overview
In this study, we first characterized the variant carrier rate (VCR), which is the proportion of individuals who carry a given variant, across a set of 415 genes previously ascribed as being associated with a severe autosomal recessive disorder.13 We calculated these VCRs using data from an exome sequencing database (gnomAD, version 2.0.2) comprised of 123,136 samples.7 Importantly, although not truly a population-based cohort, the individuals included in gnomAD have no known history of a severe Mendelian condition. These individuals were sequenced across many different sequencing platforms and at different centers, but the sequencing data underwent uniform quality control and joint variant calling.
The samples in gnomAD are distributed across seven major ancestries: African/African American (AFR, n = 7652), Hispanic (AMR, n = 16,791), Ashkenazi Jewish (ASJ, n = 4925), East Asian (EAS, n = 8624), Finnish (FIN, n = 11,150), non-Finnish European (NFE, n = 55,860), and South Asian (SAS, n = 15,391). We did not include the Finnish in this study because they comprise a small proportion of the US population. We also calculated variant frequencies for a composite US sample, which takes the variant frequencies from gnomAD and scales them by 2016 US Census data.15
We then used these VCRs to generate estimates of gene carrier rates (GCRs) for each gene, which is the estimated proportion of individuals who carry one or more pathogenic or likely pathogenic variants in that gene. Using these GCRs, we also calculated cumulative carrier rates (CCRs) for various sets of genes. These sets of genes are based on thresholds (e.g., genes having a GCR greater than a given value) and are meant to simulate hypothetical carrier screening panels. Please see Fig. 1 for a depiction of our workflow.

Scheme for paper. On top, three genes are shown: gene A (blue), B (red), and C (green). Each gene has between 2 and 4 variants, each with an associated variant carrier rate (VCR), with sample VCRs written in parentheses below (numbers represent proportions; for example, 0.001 is 0.1%). The gene carrier rate (GCR) for each gene is the probability that an individual will carry at least one variant in the given gene. Calculated GCRs based on the corresponding VCRs are in parentheses below. Finally, cumulative carrier rates (CCRs) are shown for two sample panels of genes. CCRAB (purple) is the CCR for a hypothetical panel of genes containing genes A and B. CCRABC (black) is the CCR for a hypothetical panel containing genes A, B, and C. For each hypothetical panel, the calculated CCR is in parentheses.
Carrier rates by gene
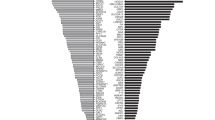
The GCR for each of the 415 genes associated with severe recessive Mendelian conditions for each of the six ancestries as well as the US composite are listed in Table S2. For illustrative purposes, Fig. 2 shows the ten highest GCRs for each of the six ancestry groups. Across ancestries, the highest GCR for a single gene is 12.0% for HBB (encoding the hemoglobin β-chain, variants in which cause hemoglobinopathies including sickle cell anemia) in AFR. In each ancestry, the carrier rates rapidly decline. For example, in ASJ, only 28 of the 415 genes had a carrier rate >1%. More strikingly, for AMR, only 5 of the 415 genes had a GCR >1%.

Gene carrier rates (GCRs) for the top ten genes for each ancestry. The genes are listed on the vertical axis, and the GCRs are shown on the horizontal axis. AFR African/African American, AMR Hispanic, ASJ Ashkenazi Jewish, EAS East Asian, NFE non-Finnish European, SAS South Asian, USA composite US.
In Figure S1, we show the top 30 genes in terms of GCR in any ancestry and show the carrier rates across ancestries for each of these genes. As can be seen, some genes such as HBB or F11 are relatively restricted to a single population (AFR and ASJ respectively). In contrast, other genes such as CYP21A2 have high GCRs across many ancestries.
Cumulative carrier rates
We ranked each gene in descending order by its GCR in the respective ancestry and plotted the CCRs as increasing numbers of genes are screened. As can be appreciated in Fig. 3a, there is initially a rapid increase in the CCR, reflecting a small number of genes with high GCRs. This is then followed by a long tail of genes that contribute asymptotically to the CCR. For example, in ASJ, 90% of the CCR is contributed by the 48 top ranking genes with the highest GCR (out of 415 total genes).

Cummulative carrier rates (CCR) across ancestries. (a) Cumulative carrier rates (CCRs) for each ancestry as additional genes are screened. Genes are ranked for each ancestry in descending order based on gene carrier rate (GCR) for that ancestry. (b) CCR when screening all 415 genes (pink), only genes with GCR >0.1% (green), >0.5% (blue), or >1.0% (purple). AFR African/African American, AMR Hispanic, ASJ Ashkenazi Jewish, EAS East Asian, NFE non-Finnish European, SAS South Asian, USAcomposite US.
We next calculated CCRs for various sets of genes, which are delineated to reflect the hypothetical construction of an ECS panel. We first examined the CCR of all 415 severe recessive genes. As can be appreciated in Fig. 3b, there were very high CCRs for this set of 415 genes in all ancestries (red bars). At the extreme, in ASJ, the CCR across all 415 genes was 62.9%. The lowest CCR for the 415 genes was in EAS at 32.6%.
ACOG recently suggested that genes with GCR >1.0% are appropriate for ECS. We thus examined how screening only genes with GCR >1% in the respective population would affect the CCR. We found that setting this >1% threshold drastically reduces the CCR by 20.6–77.8% (purple bars) (Fig. 3b). We also show CCRs if only genes with GCR >0.5% (blue bars) or >0.1% (green bars) are included (Fig. 3b). We note that these results simulate the yields that would be derived from using a hypothetical ancestry-specific ECS panel, where only genes with GCR greater than some threshold based on that ancestry are included on the panel.
Design of panethnic panels
We next examined the number of genes that meet the 1.0% GCR threshold in the respective ancestry. We found that in ASJ, 28 genes had GCR >1.0%, while in AMR, only 5 genes had GCR >1.0% (Fig. 4a, purple bars). Lowering the GCR threshold to >0.5% (blue bars) or >0.1% (green bars) greatly increases the number of genes that need to be screened.

Number of genes and CCRs for various panel designs. (a) Number of genes with gene carrier rate (GCR) >0.1% (green), >0.5% (blue), or >1.0% (purple) for each of six ancestries or for a hypothetical panethnic panel (PAN). (b–d) Cumulative carrier rate (CCR) for each ancestry using an ancestry-specific (orange) or panethnic (blue) panel comprised of genes with GCR >1.0% (b), >0.5% (c), or >0.1% (d). AFR African/African American, AMR Hispanic, ASJ Ashkenazi Jewish, EAS East Asian, NFE non-Finnish European, SAS South Asian, USA composite US.
The above analyses (Fig. 3) defined hypothetical ECS panels using GCR thresholds in each respective ancestry and are thus ancestry-specific. However, it may be desirable to apply a single panel across all ancestries (i.e., panethnic panel). We thus sought to evaluate the performance of a hypothetical panethnic panel. We calculated the number of genes that would need to comprise a panethnic panel such that all genes with a GCR greater than a threshold in any one of the individual ancestries would be included. We found that panethnic panels greatly increase the number of genes needed to be screened. For example, to include all genes with GCR >0.1% in any component ancestry, 244 genes would need to be on the panethnic panel. In contrast, there was a range of 86 (EAS) to 157 (NFE) genes with GCRs >0.1% in the individual ancestries.
We next calculated the CCR for each panethnic panel and compared it with ancestry-specific panels (Fig. 4b–d). Fig. 4b shows the CCRs for a hypothetical panethnic panel (blue bars) comprised of genes with GCR >1.0% compared with similar ancestry-specific panels (orange bars). Parallel analyses are shown for panels designed to capture genes with GCR >0.5% (Fig. 4c) and for GCR >0.1% (Fig. 4d).
At-risk couple rate
We examined the at-risk couple rate (ACR) for all 415 genes, which is the probability that both the mother and father are carriers for pathogenic or likely pathogenic variants in the same gene for at least one gene. Unsurprisingly, the highest ACRs for intra-ancestry couples was for ASJ (252 of 10,000 couples) and the lowest was for EAS (25 of 10,000 couples) (Fig. 5a). The inter-ancestry rates ranged from 17 of 10,000 EAS/AMR or EAS/SAS couples, to 76 of 10,000 NFE/ASJ couples.

At-risk couple rates (ACR) within and across ancestries. (a) Number of couples (out of 10,000) who are at risk when screening all 415 genes associated with a severe recessive condition. The ancestries are indicated on the top and the diagonal. The boxes on the diagonal show rates for couples where both individuals are of the same ancestry (intra-ancestry), and other boxes are for couples where individuals are of different ancestries (inter-ancestry). (b–d) Show similar data, except when considering a hypothetical panethnic panel comprised of genes with gene carrier rate (GCR) >1.0% (b), >0.5% (c), or >0.1% (d). AFR African/African American, AMR Hispanic, ASJ Ashkenazi Jewish, EAS East Asian, NFE non-Finnish European, SAS South Asian, USA composite US.
We also calculated the ACR when considering only genes with GCR >1.0% in any population (Fig. 5b), GCR >0.5% (Fig. 5c), or GCR >0.1% (Fig. 5d), reflecting the ACR that would result from the application of various hypothetical panethnic panels highlighted in Fig. 4. Strikingly, the yields in terms of ACR for screening just the 40 genes with GCR >1.0% are very close to the yields for screening all 415 genes (compare Fig. 5a with Fig. 5b). In fact, screening just these 40 genes would identify 76.4–96.8% of the at-risk couples that screening all 415 genes would identify.
DISCUSSION
In this study, we have leveraged a large-scale exome sequencing database to estimate the GCR for each gene associated with a severe recessive condition. We also estimated the CCRs for various sets of genes to simulate the yields of hypothetical ECS panels at various specified carrier frequency thresholds. We found very high CCRs across ancestries, including 62.9% in ASJ. We found that the proportion of at-risk couples was comparatively lower, ranging from 0.17% to 2.52% of couples.
Our study has important implications for the design of ECS panels. First, we observed that CCRs increase asymptotically as additional genes are screened. This suggests that a small number of genes accounts for the majority of carrier rates across genes, which is followed by a long tail of genes with a relatively low yield. Second, the CCRs that we observed were very high, as high as 63% in ASJ. This suggests that a large fraction of individuals who undergo PGCS will be a carrier for a pathogenic or likely pathogenic variant in a gene associated with a severe recessive Mendelian condition. This introduces the possibility of substantial cost from downstream follow-up when an individual screens positive. This underscores the need for careful consideration of which genes should be screened. Third, our study provides insight into the efficacy of panethnic panels. At more stringent carrier rate thresholds such as GCR >1.0%, constructing a panethnic panel would require relatively few additional genes as compared with an ancestry-specific panel. In contrast, at less stringent thresholds such as GCR >0.1%, a panethnic panel would require a much larger number of genes than any ancestry-specific panel. Despite much greater numbers of genes needed to be included on a panethnic panel as compared with an ancestry-specific panel, the incremental yields in terms of CCR were minimal. However, in practice, many individuals are of mixed ancestry or don’t fully know their ancestry, which could make application of ancestry-specific panels difficult.
Our paper highlights a critical insight: although the GCRs are very high for any panel of genes, the proportion of at-risk couples is comparatively much lower. For example, if all 415 genes associated with severe recessive conditions are screened in ASJ, which has the highest CCR (62.9%) among ancestries analyzed, 252 of 10,000 ASJ/ASJ couples (2.52%) would carry a likely pathogenic or pathogenic variant in the same severe recessive disease gene. For other ancestry pairings, the proportion of at-risk couples is as low as 17 of 10,000 EAS/AMR or EAS/SAS couples (0.17%). The number of affected offspring is even lower than that: assuming full penetrance, 4 to 63 per 10,000 fetuses would be affected. The ACR for couples of different ancestries (inter-ancestry) is lower than the ACR for couples of the same ancestry (intra-ancestry). In fact, in nearly all cases, the ACR for an inter-ancestry couple is lower than the ACR for either component ancestry. As an example, the ACR across all 415 genes for ASJ/ASJ couples is 2.52%, and the ACR for AFR/AFR couples is 1.9%. However, the ACR for an ASJ/AFR couple is only 0.47%. This is a reflection of how the GCRs for many genes are relatively specific to a single population (Figure S1). For example, for AFR, the top gene is HBB (associated with various hemoglobinopathies; GCR = 12.0%), but HBB has a GCR of just 0.020% in ASJ. In contrast, the top gene for ASJ is F11 (associated with factor XI deficiency; GCR = 8.1%), but F11 has a GCR of just 0.010% in AFR. For these genes where the GCR is relatively ancestry-specific, the rate of both individuals of a couple with discordant ancestries each carrying a variant will be very low. However, in some inter-ancestry couples, the inter-ancestry ACR is actually higher than the intra-ancestry ACR of one of the component ancestries. This occurs in inter-ancestry couples where one individual in the couple is from a higher CCR ancestry (e.g., AFR or ASJ) and tends to elevate the predicted ACR when mating with an individual from a lower CCR ancestry (e.g., EAS or SAS).
Strikingly, we found that screening just a small handful of genes captures the vast majority of the at-risk couples that would be identified screening all 415 genes. For example, for an ASJ/ASJ couple, 2.52% of couples would be identified as being as at-risk when screening all 415 genes. However, screening just the 40 genes with GCR >1.0% in any ancestry would identify 2.41% of couples as being at risk. Screening the 87 genes with GCR >0.5% would identify 2.50% of couples, and screening the 244 genes with GCR >0.1% would identify nearly all 2.52% of at-risk couples. This suggests that lowering the threshold for genes that should be screened greatly increases the number of genes that would need to be screened, but results in a modest increase in the CCR and a minuscule increase in the ACR. However, given that the technical cost of adding additional genes is becoming increasingly lower, the balance may in fact favor including more genes on a screening panel, as genes with low GCRs (and thus lower yields) will also not incur high rates of follow-up costs.
Other studies have also examined carrier rates for recessive conditions. Haque et al. performed similar analyses for a commercial ECS panel. However, their study was limited in that it only examined 94 genes.12 Also, the majority of the samples in that study were analyzed using targeted genotyping panels that assayed only 417 preselected variants, and thus were not comprehensive in terms of the variants examined in the targeted genes. Other studies have been more comprehensive in terms of variants and genes analyzed, but have been limited to only a few hundred individuals analyzed.16,17,18 Thus, our study provides a comprehensive and large-scale examination of carrier rates for many genes associated with recessive disorders.
There are several important limitations to our study. First, we only examined coding variants in autosomal recessive genes. Our study cannot be extended to genes acting through alternative modes of inheritance, such as X-linked recessive. We also could not reliably examine more complex forms of genetic variation, such as copy-number variants (CNVs; e.g., deletions in SMN1 causing spinal muscular atrophy).19 Therefore, our data underestimate carrier rates for some genes. Second, characterizing the severity of disorders associated with genes in determining appropriateness for screening is a challenging task. Here, we used a previously published list of 415 genes associated with recessive conditions that were ascribed as being severe.13 However, what comprises a condition worthy of screening in terms of phenotypic features is a challenging technical and ethical question that is beyond the scope of this work. Other papers have begun to examine this question, but this question remains subject to debate.20 As a related issue, many of these genes might have lower penetrance and/or variable expressivity and therefore might not be appropriate for screening. Third, we chose to only include variants annotated as likely pathogenic or pathogenic in ClinVar.8,9,11 Variants were also not filtered based on their review status, as many variants in ClinVar have not achieved high levels of review status yet. Some of the variants included in this study are likely actually benign, while there may be other variants that we did not include that are actually pathogenic.21,22 This issue of variant selection is well beyond the scope of this work, although in actual practice, the variants included on commercial screening panels are subject to similar challenges in variant selection. Finally, our work examined the probability of an individual being a carrier for at least one variant, and we did not explicitly examine the probability of individuals being carriers for more than one variant. Our calculations also assume independence among variants, an assumption that may easily be violated in the context of assortative mating or inbreeding.
Another caveat of our study is that gnomAD might not truly reflect the carrier rates of the general population or of individuals who seek carrier screening, despite the fact that individuals with known severe pediatric diseases and their first-degree relatives were excluded from gnomAD.7 gnomAD likely has biased socioeconomic and ancestry breakdown (for example, many individuals in gnomAD are from relatively small population groups such as Sweden or Taiwan) when compared with the general US population or those who would undergo ECS. Many individuals in gnomAD were from cohort studies aimed at studying specific diseases such as epilepsy. Additionally, although gnomAD excluded individuals with severe pediatric diseases and their close relatives, gnomAD may still be enriched for variant carriers in more distant relatives for some genes. This is counterbalanced by the possibility that by excluding individuals with severe pediatric diseases and their close relatives, gnomAD may actually be underestimating carrier rates for some genes associated with diseases that were under study in the component cohorts in gnomAD. These effects are very difficult to quantify, but should be recognized as an important caveat to our results.
In summary, our work informs the selection of genes that should be considered for screening in PGCS and provides an important foundation for policy makers as they consider the implications of panethnic application of PGCS panels. Our data will also facilitate counseling of patients as they consider PGCS.
References
Gregg AR. Expanded carrier screening. Obstet Gynecol Clin North Am. 2018;45:103–112.
Edwards JG, Feldman G, Goldberg J, et al. Expanded carrier screening in reproductive medicine—points to consider. Obstet Gynecol. 2015;125:653–662.
Wetterstrand K. DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP). https://www.genome.gov/27541954/dna-sequencing-costs-data. Accessed 7 September 2018.
Green R, Roberts S, Cupples A, et al. Disclosure of APOE genotype for risk of Alzheimer’s disease. N Engl J Med. 2009;361:245–254.
Stewart KFJ, Wesselius A, Schreurs MAC, Schols AMWJ, Zeegers MP. Behavioural changes, sharing behaviour and psychological responses after receiving direct-to-consumer genetic test results: a systematic review and meta-analysis. J Community Genet. 2018;9:1–18.
Committee opinion no. 690 summary: carrier screening in the age of genomic medicine. Obstet Gynecol. 2017;129:595–596.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067.
Rehm HL, Berg JS, Brooks LD, et al. ClinGen—the Clinical Genome Resource. N Engl J Med. 2015;372:2235–2242.
Zhang X, Minikel EV, O’Donnell-Luria AH, MacArthur DG, Ware JS, Weisburd B. ClinVar data parsing. Wellcome Open Res. 2017;2:33.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Haque IS, Lazarin GA, Kang HP, Evans EA, Goldberg JD, Wapner RJ. Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA. 2016;316:734.
Tarailo-Graovac M, Zhu JYA, Matthews A, van Karnebeek CDM, Wasserman WW. Assessment of the ExAC data set for the presence of individuals with pathogenic genotypes implicated in severe Mendelian pediatric disorders. Genet Med. 2017;19:1300–1308.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993.
US Census. http://factfinder.census.gov/faces/nav/jsf/pages/index.xhtml. Accessed 1 March 2018.
Punj S, Akkari Y, Huang J, et al. Preconception carrier screening by genome sequencing: results from the clinical laboratory. Am J Hum Genet. 2018;0:1–12.
Arjunan A, Litwack K, Collins N, Charrow J. Carrier screening in the era of expanding genetic technology. Genet Med. 2016;18:1214–1217.
Baskovich B, Hiraki S, Upadhyay K, et al. Expanded genetic screening panel for the Ashkenazi Jewish population. Genet Med. 2016;18:522–528.
Prior TW, Professional Practice and Guidelines Committee. Carrier screening for spinal muscular atrophy. Genet Med. 2008;10:840–842.
Beauchamp KA, Muzzey D, Wong KK, et al. Systematic design and comparison of expanded carrier screening panels. Genet Med. 2018;20:55–63.
Yang S, Lincoln SE, Kobayashi Y, Nykamp K, Nussbaum RL, Topper S. Sources of discordance among germ-line variant classifications in ClinVar. Genet Med. 2017;19:1118–1126.
Amendola LM, Jarvik GP, Leo MC, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98:1067–1076.
Acknowledgements
We thank the individuals who contributed their samples to the gnomAD database, as well as the gnomAD team for compiling the resource and sharing it with the world to use.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
A.R.G. is on the Board of ACMG and the ACMG Foundation. M.H.G. declares no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Guo, M.H., Gregg, A.R. Estimating yields of prenatal carrier screening and implications for design of expanded carrier screening panels. Genet Med 21, 1940–1947 (2019). https://doi.org/10.1038/s41436-019-0472-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0472-7
Keywords
This article is cited by
-
Prevalence of common autosomal recessive mutation carriers in women in the Southern Vietnam following the application of expanded carrier screening
Scientific Reports (2024)
-
Miscarriage risk assessment: a bioinformatic approach to identifying candidate lethal genes and variants
Human Genetics (2024)
-
Scaling-up and future sustainability of a national reproductive genetic carrier screening program
npj Genomic Medicine (2023)
-
Populational pan-ethnic screening panel enabled by deep whole genome sequencing
npj Genomic Medicine (2023)
-
Carrier burden of over 300 diseases in Han Chinese identified by expanded carrier testing of 300 couples using assisted reproductive technology
Journal of Assisted Reproduction and Genetics (2023)
