
Abstract
Purpose
The interpretation of genetic variants after genome-wide analysis is complex in heterogeneous disorders such as intellectual disability (ID). We investigate whether algorithms can be used to detect if a facial gestalt is present for three novel ID syndromes and if these techniques can help interpret variants of uncertain significance.
Methods
Facial features were extracted from photos of ID patients harboring a pathogenic variant in three novel ID genes (PACS1, PPM1D, and PHIP) using algorithms that model human facial dysmorphism, and facial recognition. The resulting features were combined into a hybrid model to compare the three cohorts against a background ID population.
Results
We validated our model using images from 71 individuals with Koolen–de Vries syndrome, and then show that facial gestalts are present for individuals with a pathogenic variant in PACS1 (p = 8 × 10−4), PPM1D (p = 4.65 × 10−2), and PHIP (p = 6.3 × 10−3). Moreover, two individuals with a de novo missense variant of uncertain significance in PHIP have significant similarity to the expected facial phenotype of PHIP patients (p < 1.52 × 10−2).
Conclusion
Our results show that analysis of facial photos can be used to detect previously unknown facial gestalts for novel ID syndromes, which will facilitate both clinical and molecular diagnosis of rare and novel syndromes.
Similar content being viewed by others

INTRODUCTION
Rare sporadic (de novo) pathogenic variants are a major cause of developmental disorders such as intellectual disability (ID) and autism spectrum disorders.1 Using next-generation sequencing strategies, a disease-causing genetic variant can now be identified in ~60% of individuals with ID, and to date approximately 750 of the predicted 2000 genes causing ID have been identified.1,2,3 In 30–40% of cases, ID is observed as part of a broader syndrome consisting of facial dysmorphology in conjunction with additional congenital abnormalities.4 In addition, we have previously shown that individuals with ID who present with facial dysmorphology are significantly enriched in both pathogenic and likely pathogenic variants.5 Despite the distinctive, although sometimes subtle facial features, phenotyping of individuals with ID (and their parents) currently relies on a clinician’s ability to recognize a syndrome based on its related dysmorphology.
The implementation of high-throughput sequencing technologies such as exome sequencing has facilitated the ability to detect individuals with increasingly rare novel disorders;2 this calls for novel methods for collecting phenotypic information. Facial recognition and matching algorithms have matured in recent years and can be exploited in a clinical setting to automate and objectively measure a person’s dysmorphology with minimal impact on the person.6,7,8,9,10,11,12,13,14,15,16,17,18 The advantages of these methods are that they also include subtle features that may be difficult for clinicians to identify or recognize features that may not necessarily be considered dysmorphic. Moreover, they can be used to identify additional individuals with a known syndrome or aid the characterization of novel ID syndromes. Alternatively, they can support the interpretation of variants for which functional impact may be uncertain, such as missense variants in a gene known to cause disease when mutated but for which clinical presentation is variable and/or includes a broad clinical spectrum. In this study, we created a novel hybrid learning model for detecting facial similarity within small subsets of individuals with the same ID syndrome (n < 15) and compared them with the general ID population. Our new model was able to successfully detect facial similarity in all three examined novel ID syndromes by incorporating both dysmorphic and generic facial features from two different facial analysis algorithms in combination with a unique collection of photos of control individuals with ID.12,19 We show that our new hybrid model is more sensitive than individual facial analysis algorithms and that it can lead to novel clinical diagnosis for individuals with missense variants of uncertain significance.
MATERIALS AND METHODS
Data collection
For this study one validation set and three test sets were generated. Each set consisted of facial photos of individuals with a specific ID syndrome and of matched ID controls. All facial photographs used in the study were taken from an approximately frontal position under uncontrolled conditions. The majority of the individuals with a specific ID syndrome used in this study are previously published cases. In addition some novel unpublished cases were included. The individuals selected as ID controls are previously unpublished individuals who were enrolled in the study from routine clinical diagnostics at the Department of Human Genetics at the Radboud University Medical Center (Radboudumc). To ensure anonymity all photographs were converted into unidentifiable feature vectors before analysis. This study was approved by the RadboudUMC under the realm of diagnostic process.
Validation set
The validation set consisted of 71 facial photos of Caucasian individuals with Koolen–de Vries syndrome (KdVS) (OMIM ID 610443), a syndrome with relatively high number of patients and a known facial gestalt. For each patient, one ID control was selected matching in gender, ethnicity, and age (measured in years) at the time of the photo. This resulted in a validation set consisting of 71 individuals with KdVS and 71 matched ID controls. (Supplemental Table S1).
Test sets
Three test sets were collected, each containing photos of Caucasian individuals with variants in a specific novel ID gene (PACS1 [Schuurs–Hoeijmakers syndrome, OMIM 615009; N = 14], PPM1D [OMIM 617450; N = 11], and PHIP [OMIM 612870; N = 16]) and matched ID controls. For each patient five ID controls were selected matching in gender, ethnicity, and age (measured in years) at the time of the photo. This resulted in three separate test sets: (1) PACS1 test set consisting of 14 individuals with Schuurs–Hoeijmakers syndrome and 70 matched ID controls, (2) a PPM1D test set consisting of 11 individuals with a pathogenic truncating variant in the last or penultimate exon of PPM1D with 55 matched ID controls, and (3) a PHIP test set consisting of 16 individuals with a pathogenic variant in PHIP and 80 matched ID controls (Supplemental Table S2–S4).
The PACS1 individuals all had the identical pathogenic de novo missense variant in PACS1 (NM_018026.2:c.607 C>T; p.[Arg203Trp]),20,21 the 11 PPM1D individuals all had pathogenic truncating variants in the last or penultimate exon of PPM1D (NM_003620.3) (ref. 22), whereas the individuals with variants in PHIP (NM_017934.6) formed a more heterogeneous population, consisting of frameshifts, nonsense, splice site, and missense variants.23 The four PHIP missense variant (of uncertain significance) patients and their controls were excluded from the gestalt analysis of PHIP patients and were separately analyzed in a later stage, to test if their faces match the PHIP facial phenotype.
ID control population
The control population consisted of ID patients referred to the clinical genetics center of the RadboudUMC, Nijmegen due to unexplained ID without a clearly recognizable syndromic form of ID. From these ID patients, 15% had a definitive genetic diagnosis, a possible cause was identified in 24% of cases (e.g., a variant of uncertain significance), and for 61% of the control cohort the genetic cause remained unknown. Instead of using healthy control, ID controls were used to examine if we can distinguish a specific ID syndrome from the general ID population on the basis of facial features. As described above, controls were selected to match the patient in gender, ethnicity, and age (measured in years) at the time of the photo. Of all patients in the validation and test sets, 86% had corresponding controls that were an exact age match, whereas for the remaining 14% perfect age matches were not available and the closest available ages were selected (Supplemental Table S1–S4). Patients were excluded from the study if the age difference between patient and control was greater than one-third of the patient’s age.
Feature extraction
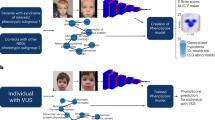
A hybrid face description vector was extracted for each portrait photo by combining the output of two computer vision algorithms: the Clinical Face Phenotype Space (CFPS)12 and OpenFace.19 Both these pipelines analyze a portrait photo to extract a feature vector describing the facial characteristics. In brief, the CFPS pipeline12 first uses face detection, landmark annotation, and shape and appearance extraction to obtain a face representation and transforms this face representation into a 340-dimensional feature vector that represents the patient’s facial features. The resulting space places two individuals closer together if they have similar dysmorphic features and farther apart if they do not. The OpenFace pipeline19 is a system designed for facial recognition. It uses face detection, landmark annotation, and affine transformation to convert a portrait photo into a standardized 96 × 96 pixel image. This image is then used as input for a pretrained facial recognition deep neural network of which the output is a 128-dimensional feature vector that describes the facial characteristics relevant to facial recognition. It creates an abstract space in which people with similar faces are located closer together, while people with dissimilar faces are located farther apart.
We applied both the CFPS and OpenFace pipeline to our KdVS, PACS1, PPM1D, and PHIP data sets, resulting in two feature vectors for each photo. Because both feature extraction methods are trained for a different purpose they are sensitive to different facial features. The two feature vectors are then combined into one hybrid vector, thus incorporating dysmorphic information as well as general facial characteristics. Before combining the vectors, normalization was applied to give both models an equal weight. The mathematical expression for the resulting hybrid feature vector is described by
Where fO is the 128-dimensional OpenFace feature vector, fC is the 340-dimensional CFPS feature vector, and \(\left\| z \right\|_2\) is the Euclidean norm of z. This process results in one 468-dimensional hybrid feature vector describing the facial features for each individual in the data sets.
Gestalt analysis
The similarity between patients with the same novel ID syndrome was analyzed by calculating the clustering improvement factor (CIF) for each of the four novel ID syndromes, using the patients as positives and the controls as negatives. The CIF gives an estimate of the search space reduction.12 In summary, the CIF estimates how well a group of positives (e.g., patients) cluster within a group of negatives (e.g., the controls) compared with what would be expected by random chance. The CIF is calculated by determining for each positive (e.g., patient), the rank of the nearest other positive (e.g., patient) and then comparing the average of these observed ranks O(r) to the expected rank of the nearest other positive E(r). And described by
Where the expected rank is described as
And where Nn is the number of negative instances and Np is the number of positive instances.
After determining the CIF for each syndrome we randomly labeled (while maintaining the patient–control ratio) the patients and controls in each data set 10,000 times and calculated the corresponding CIF for every permutation. For each syndrome a right-tailed Mann–Whitney U test was performed to determine if the CIF for the syndrome is significantly higher than expected by random chance. This process is performed for each of the three feature types: CFPS, OpenFace, and hybrid. We also compared the performance of these feature types on smaller numbers of patients. For this purpose, we calculated the CIF for each possible combination of N patients in each test set. Then we compared the median CIF of all possible combinations with the CIFs for 1000 random permutations for each N as described above. In addition, the distribution of the hybrid feature vectors of the patients and controls are visualized for each data sets using t-distributed stochastic neighbor embedding (t-SNE) to reduce dimensionality.
Additionally, an experiment was performed to evaluate the effect that the number of patients versus controls had on the resulting CIF and p values. For each test set the corresponding controls were divided into five disjoint subsets resulting in subsets containing one matched ID control for each patient in the test set, thus matching the patient-to-control ratio used in the validation set. Then across each syndrome the CIF and p value were calculated for each of the five control subsets using the hybrid model and evaluated via the mean p value and resulting confidence interval.
RESULTS
To examine whether a similar facial gestalt is present within three novel ID syndromes using computational analysis, we propose a descriptive facial feature vector that combines the OpenFace and CFPS feature vector. Next, we calculated the CIF to detect significant similarity. The CIF is a factor that estimates how well the patients cluster within the group of controls.
Model validation using a known syndrome
We first validated our model on patients with KdVS syndrome, which is known to have associated and consistent facial dysmorphology, including a long hypotonic face and a bulbous nasal tip.24 The CFPS, OpenFace, and hybrid feature vectors were extracted and compared between the 71 KdVS patients and their age- and gender-matched controls. The faces of the KdVS patients had a higher CIF than expected by random chance, both when using the CFPS (CIF = 0.993, p = 4.26 × 10−2) and the OpenFace (CIF = 1.143, p = 2 × 10−4) features, meaning both feature types can detect the facial similarity in KdVS patients. Our novel hybrid feature vector also showed a higher CIF than expected by random chance (CIF = 1.191, p < 1 × 10−4). The CIF for the hybrid model is higher than for the individual feature types, indicating an improvement in clustering when combining the two feature vectors (Table 1).
Model application to novel ID syndromes
We performed a similar analysis for the three novel ID syndromes, with five age-matched controls per patient, using either the CFPS, OpenFace, or our novel hybrid model to describe the facial characteristics. Using only the CFPS features significant similarity was detected for PACS1 (CIF = 1.908, p = 1.90 × 10−2) and PHIP (CIF = 2.399, p = 7.1 × 10−3), but not for PPM1D (CIF = 1.011, p = 4.77 × 10−1). Using the OpenFace features found significant similarity for PACS1 (CIF = 1.955, p = 7.6 × 10−3) and PPM1D (CIF = 1.667, p = 4.80 × 10−2), but not for PHIP (CIF = 1.101, p = 3.20 × 10−1). Our novel hybrid model was the only model that showed significant facial similarity for all three syndromes: PACS1 (CIF = 2.523, p = 8 × 10−4), PPM1D (CIF = 1.713, p = 4.65 × 10−2), and PHIP (CIF = 2.239, p = 6.3 × 10−3) (Table 1). For all three novel ID genes the highest significance was achieved when using the hybrid feature vectors. A t-SNE plot of the different test sets illustrates the similarity between the patients with the same syndrome compared with the matched ID control population using our novel hybrid model (Fig. 1).

Distribution of the hybrid facial features in the PACS1, PPM1D, and PHIP data sets. The t-distributed stochastic neighbor embedding (t-SNE) plots of the (a) PACS1, (b) PPM1D, and (c) PHIP data set analyzed using our novel hybrid model show that faces of patients with the same novel intellectual disability (ID) syndrome are located close together within a group of age-, gender-, and ethnicity-matched controls. Four individuals with a missense variant of uncertain significance in the PHIP gene are compared with 12 individuals with a presumed loss-of-function variant in PHIP, showing significant similarity to the PHIP facial phenotype for individuals A and D.
Application to missense variants of uncertain significance
We examined whether the four patients with a de novo missense variant in the PHIP gene (individual A: NM_017934.6:c.328C>T, individual B: NM_017934.6:c.328C>A, individual C: NM_017934.6:c.1562A>G, and individual D: NM_017934.6:c.2888A>G) show significant similarity to the 12 individuals with a presumed loss-of-function variant in PHIP. When visualizing the complete PHIP data set, we observed that all four missense patients were located in the vicinity of the other PHIP patients. Individuals A and D displayed a stronger association to other PHIP patients compared with individuals B and C (Fig. 1c). To quantify this, we compared the Euclidean distance from the hybrid feature vectors of the missense patients with their nearest PHIP patient with the Euclidean distances between the controls and their nearest PHIP patient using a right-tailed Mann–Whitney U test (Table 2). The distance from individuals A and D to the nearest PHIP patient was significantly smaller than expected by random chance (both p < 1.52 × 10−2), thus showing that the facial phenotypes of individuals A and D are significantly similar to the facial phenotype of other PHIP patients. Whereas individuals B and C were not significantly closer to other PHIP patients than expected by random chance (p = 1.82 × 10−1 and p = 6.06 × 10−2 respectively).
DISCUSSION
The development of the face and the brain are tightly linked and craniofacial anomalies are reported in 30–40% of individuals with ID.4 This link has been used in clinical setting for decades to establish a syndromic diagnosis in individuals with ID. Developments in facial phenotyping through computational analysis of facial images have shown that it is possible to train an algorithm to recognize patients with a number of known dysmorphic syndromes.6,7,8,9,10,11,12,13,14,15,16,17,18 In this study we used facial image analysis to answer two different questions, namely (1) is there a shared facial gestalt present for a novel ID syndrome without clear clinically recognizable facial features, and (2) do patients with a variant of uncertain significance exhibit significant facial similarity to patients with a pathogenic variant within the same gene. To answer these questions, in contrast to previous studies, we used a background control set of individuals with ID, and not healthy individuals. Thereby presenting the algorithm with a relevant classification problem close to the clinical question, namely to recognize subgroups of individuals within the ID population. In addition, we matched the controls to exclude age-, gender-, or ethnicity biases.
Information contained in human faces can be used in the delineation of genetic entities.18 For this reason, we selected four different cohorts of individuals with syndromes caused by different pathophysiological mechanisms. Firstly, for validation purposes we selected a large cohort of KdVS patients representing a homogeneous set of patients with loss-of-function variants correlating to a known syndrome with dysmorphic features. Our model also showed that the faces of individuals with KdVS were significantly similar when compared with the general ID population. Similarly the patients with a de novo PACS1 variant represent a cohort in which all members had an identical pathogenic de novo variant in PACS1 (NM_018026.2:c.607C>T; p.[Arg203Trp])20,21 and for which computational analysis had recently been performed on a subset of the individuals showing that a facial gestalt is present.12,16 Notably in comparison with the first two index patients, the facial phenotype has broadened as the cohort has expanded, and more variation has been introduced while still showing that significant facial similarity exists between individuals (Supplemental Figure S1). Our model achieved higher CIF values for the test cohorts compared with the validation cohort, in part due to the difference in patient-to-control ratio, which results in a higher maximum achievable CIF. Retrospective analysis illustrated that selecting one matched ID control per test subject influenced the resulting CIF values and had insufficient power to generate statistically robust results (Supplemental Table S5). The last two cohorts consist of patients without a clearly clinically recognizable facial dysmorphology; PPM1D patients all had pathogenic truncating variants in the last or penultimate exon of PPM1D (NM_003620.3) (ref. 22) and the cohort of PHIP patients had variants (NM_017934.6) consisting of loss-of-function variants (frameshifts, nonsense, splice sites) leading to haploinsufficiency.23 This final cohort of PHIP patients allowed us to test the ability to classify patients with a de novo missense variant of uncertain significance within the clinical context of the remainder of PHIP patients with a presumed loss-of-function variant. The pathophysiological effect of a missense variant is often less clear than that of truncating variants. Our analysis helped interpret the phenotypic effect of two of four missense variants, demonstrating in two individuals (A and D) significant similarity to other PHIP patients with a loss-of-function variant. Whereas individuals B and C showed a trend toward facial similarity, which may become significant with a larger cohort of PHIP individuals.
While choosing not to retrain the algorithms, our model does combine the CFPS feature space trained to recognize dysmorphic features in patients with the features from OpenFace trained to perform face recognition. We show that the two feature spaces are sensitive to different characteristics and only their combined power was able to detect the sometimes-subtle features within our small patient cohorts (Supplemental Figure S2). For example, CFPS detected similarity in the PHIP patient group, whereas OpenFace detected similarity in the PPM1D patient group. This most likely reflects the more distinct facial features of individuals with a pathogenic variant in PHIP compared with the subtler facial features of individuals with a pathogenic PPM1D variant. The disadvantage of both models is that it results in an abstract representation of the face and it is difficult to translate the results of the classification back to individual facial features and hence underlying biology. However, computer-based models do perform objective facial analysis, compared with the more subjective interpretations of clinicians. One challenge that needs to be faced in future research is the comparison of facial features between patients with different ethnicities.25,26,27,28,29
In conclusion we present a model that combines a facial recognition algorithm with an algorithm trained to recognize dysmorphic features. We show that patients with a pathogenic variant in one of three novel ID genes (PACS1, PPM1D, and PHIP), identified via genotype-first approach, have an associated facial gestalt, thus demonstrating that information contained in the face can be used to delineate genetic entities including in novel ID syndromes with no previously known knowledge of a facial phenotype. Notably our model provides significant results with a small number of patients (n < 15) and provides a phenotypic readout to interpret variants of uncertain significance. The implementation of next-generation sequencing in the clinic has resulted in the identification of increasingly rare syndromes, and the orthogonal use of computational analysis of facial features will be key for linking variants to patients’ novel and yet unknown phenotypes leading to a clinical diagnosis.
URLs
CADD http://cadd.gs.washington.edu/ Clinical Face Phenotype Space https://github.com/ChristofferNellaker/Clinical_Face_Phenotype_Space_Pipeline OpenFace https://cmusatyalab.github.io/openface/
References
Gilissen C, Hehir-Kwa JY, Thung DT, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344.
Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17:9–18.
Chiurazzi P, Pirozzi F. Advances in understanding–genetic basis of intellectual disability. F1000Research. 2016;5:599.
Hart TC, Hart PS. Genetic studies of craniofacial anomalies: clinical implications and applications. Orthod Craniofac Res. 2009;12:212–220.
Vulto‐van Silfhout AT, Hehir‐Kwa JY, Bon BW, et al. Clinical significance of de novo and inherited copy‐number variation. Hum Mutat. 2013;34:1679–1687.
Loos HS, Wieczorek D, Wurtz RP, von der Malsburg C, Horsthemke B. Computer-based recognition of dysmorphic faces. Eur J Hum Genet. 2003;11:555–560.
Hammond P, Hutton TJ, Allanson JE, et al. Discriminating power of localized three-dimensional facial morphology. Am J Hum Genet. 2005;77:999–1010.
Boehringer S, Vollmar T, Tasse C, et al. Syndrome identification based on 2D analysis software. Eur J Hum Genet. 2006;14:1082–1089.
Dalal AB, Phadke SR. Morphometric analysis of face in dysmorphology. Comput Methods Programs Biomed. 2007;85:165–172.
Vollmar T, Maus B, Wurtz RP, et al. Impact of geometry and viewing angle on classification accuracy of 2D based analysis of dysmorphic faces. Eur J Med Genet. 2008;51:44–53.
Boehringer S, Guenther M, Sinigerova S, Wurtz RP, Horsthemke B, Wieczorek D. Automated syndrome detection in a set of clinical facial photographs. Am J Med Genet A. 2011;155A:2161–2169.
Ferry Q, Steinberg J, Webber C, et al. Diagnostically relevant facial gestalt information from ordinary photos. eLife. 2014;3:e02020.
Balliu B, Wurtz RP, Horsthemke B, Wieczorek D, Bohringer S. Classification and visualization based on derived image features: application to genetic syndromes. PLoS One. 2014;9:e109033.
Wang K, Luo J. Detecting visually observable disease symptoms from faces. EURASIP J Bioinform Syst Biol. 2016;2016:13.
Cerrolaza JJ, Porras AR, Mansoor A, Zhao Q, Summar M, Linguraru MG. Identification of dysmorphic syndromes using landmark-specific local texture descriptors. Paper presented at: IEEE 13th International Symposium on Biomedical Imaging (ISBI). April 13–16, 2016; Prague, Czech Republic.
Dudding-Byth T, Baxter A, Holliday EG, et al. Computer face-matching technology using two-dimensional photographs accurately matches the facial gestalt of unrelated individuals with the same syndromic form of intellectual disability. BMC Biotechnol. 2017;17:90.
Liehr T, Acquarola N, Pyle K, et al. Next generation phenotyping in Emanuel and Pallister Killian syndrome using computer-aided facial dysmorphology analysis of 2D photos. Clin Genet. 2018;93:378–381.
Knaus A, Pantel JT, Pendziwiat M, et al. Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med. 2018;10:3.
Amos B, Ludwiczuk B, Satyanarayanan M. Openface: A general-purpose face recognition library with mobile applications. CMU School of Computer Science. 2016.
Schuurs-Hoeijmakers JH, Oh EC, Vissers LE, et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am J Hum Genet. 2012;91:1122–1127.
Schuurs-Hoeijmakers JH, Landsverk ML, Foulds N, et al. Clinical delineation of the PACS1-related syndrome—report on 19 patients. Am J Med Genet A. 2016;170:670–675.
Jansen S, Geuer S, Pfundt R, et al. De novo truncating mutations in the last and penultimate exons of PPM1D cause an intellectual disability syndrome. Am J Hum Genet. 2017;100:650–658.
Jansen S, Hoischen A, Coe BP, et al. A genotype-first approach identifies an intellectual disability-overweight syndrome caused by PHIP haploinsufficiency. Eur J Hum Genet. 2018;26:54–63.
Koolen DA, Vissers LE, Pfundt R, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38:999–1001.
Lumaka A, Cosemans N, Lulebo Mampasi A, et al. Facial dysmorphism is influenced by ethnic background of the patient and of the evaluator. Clin Genet. 2017;92:166–171.
Kruszka P, Addissie YA, McGinn DE, et al. 22q11.2 deletion syndrome in diverse populations. Am J Med Genet A. 2017;173:879–888.
Kruszka P, Porras AR, Addissie YA, et al. Noonan syndrome in diverse populations. Am J Med Genet A. 2017;173:2323–2334.
Kruszka P, Porras AR, Sobering AK, et al. Down syndrome in diverse populations. Am J Med Genet A. 2017;173:42–53.
Kruszka P, Porras AR, de Souza DH, et al. Williams-Beuren syndrome in diverse populations. Am J Med Genet A. 2018;176:1128–1136.
Acknowledgements
We thank the individuals and their parents for participating in this study. We thank the clinicians for referring cases. We thank Milan Baars, Alexander Currie, and Martijn Mandigers for their contribution in collecting patient photographs. This work was financially supported by grants from the Netherlands Organization for Scientific Research (NWO) (916-16-015 to J.Y.H.-K. and 912-12-109 to L.E.L.M.V. and B.B.A.d.V.). The Medical Research Council (UK) has funded C.N. through a MRC Methodology Research Fellowship MR/M014568/1. The patient images used in this study cannot be made openly available due to patient privacy concerns. Please contact the authors with specific queries regarding data access.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
van der Donk, R., Jansen, S., Schuurs-Hoeijmakers, J.H.M. et al. Next-generation phenotyping using computer vision algorithms in rare genomic neurodevelopmental disorders. Genet Med 21, 1719–1725 (2019). https://doi.org/10.1038/s41436-018-0404-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0404-y
Keywords
This article is cited by
-
Integrative analysis of transcriptome dynamics during human craniofacial development identifies candidate disease genes
Nature Communications (2023)
-
PhenoScore quantifies phenotypic variation for rare genetic diseases by combining facial analysis with other clinical features using a machine-learning framework
Nature Genetics (2023)
-
Using deep-neural-network-driven facial recognition to identify distinct Kabuki syndrome 1 and 2 gestalt
European Journal of Human Genetics (2022)
-
Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON
European Journal of Human Genetics (2022)
-
GestaltMatcher facilitates rare disease matching using facial phenotype descriptors
Nature Genetics (2022)
